From the Indiana University School of Medicine, Indianapolis (S.C., S.K.G., X.C., B.F.M., T.M.C., J.K., A.G.O., M.C.).
Notre Dame University, South Bend, IN (B.W.E., P.Z.).
Circ Res. 2019 Oct 11;125(9):805-820. doi: 10.1161/CIRCRESAHA.119.315082. Epub 2019 Aug 27.
Even in antiretroviral therapy-treated patients, HIV continues to play a pathogenic role in cardiovascular diseases. A possible cofactor may be persistence of the early HIV response gene Nef, which we have demonstrated recently to persist in the lungs of HIV+ patients on antiretroviral therapy. Previously, we have reported that HIV strains with Nef, but not Nef-deleted HIV strains, cause endothelial proinflammatory activation and apoptosis.
To characterize mechanisms through which HIV-Nef leads to the development of cardiovascular diseases using ex vivo tissue culture approaches as well as interventional experiments in transgenic murine models.
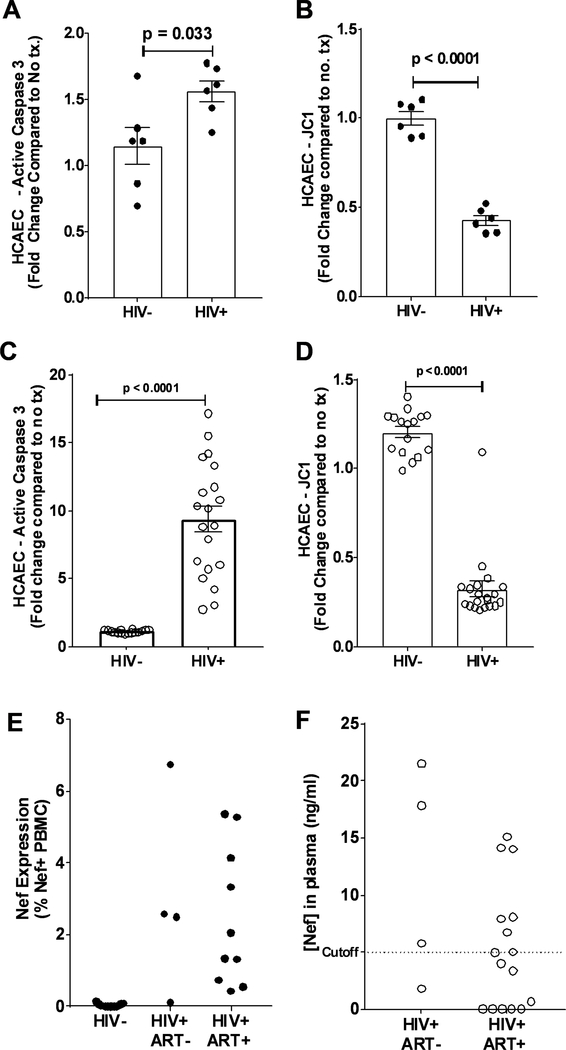
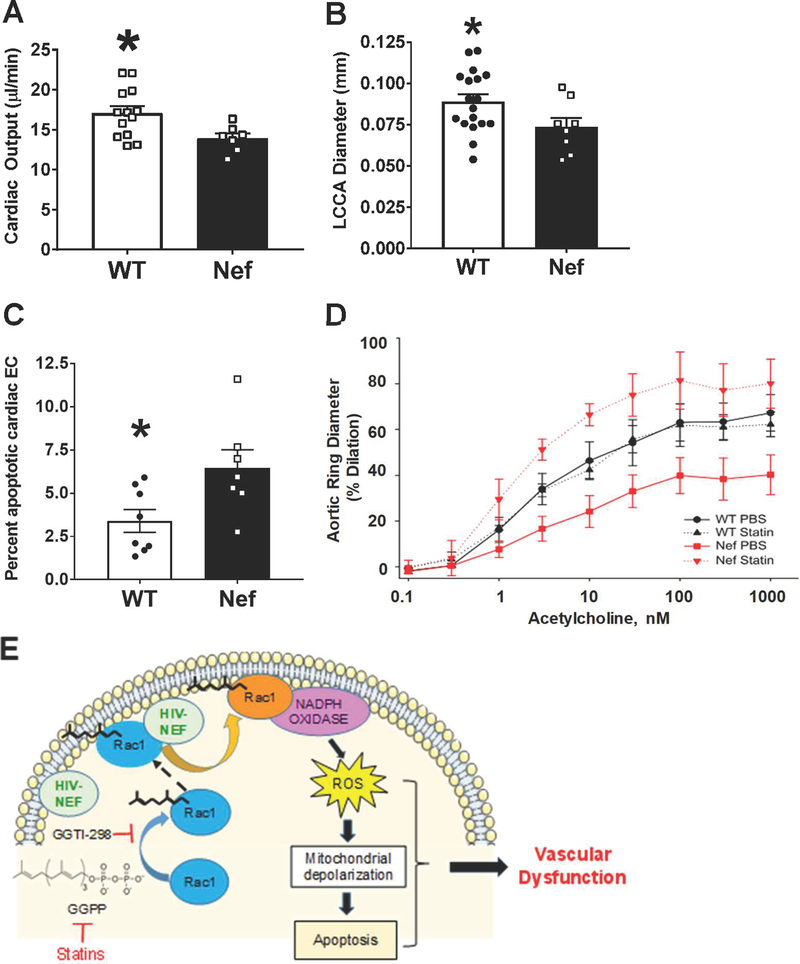
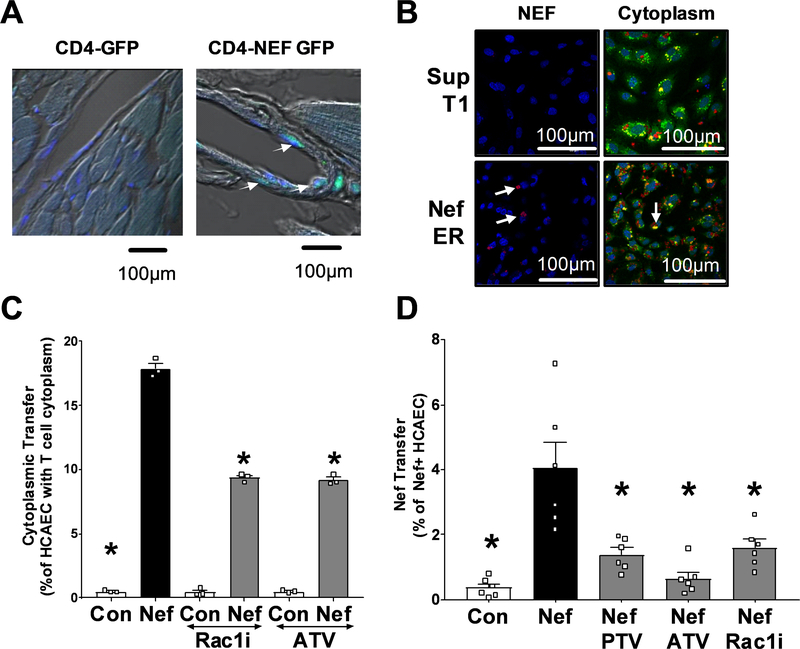
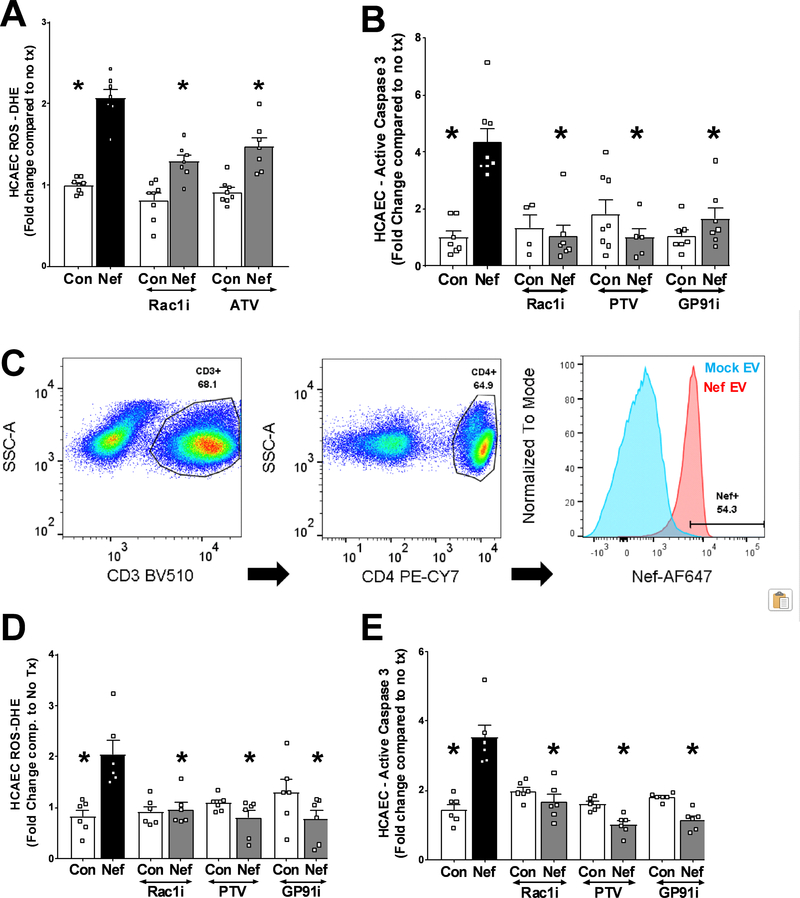
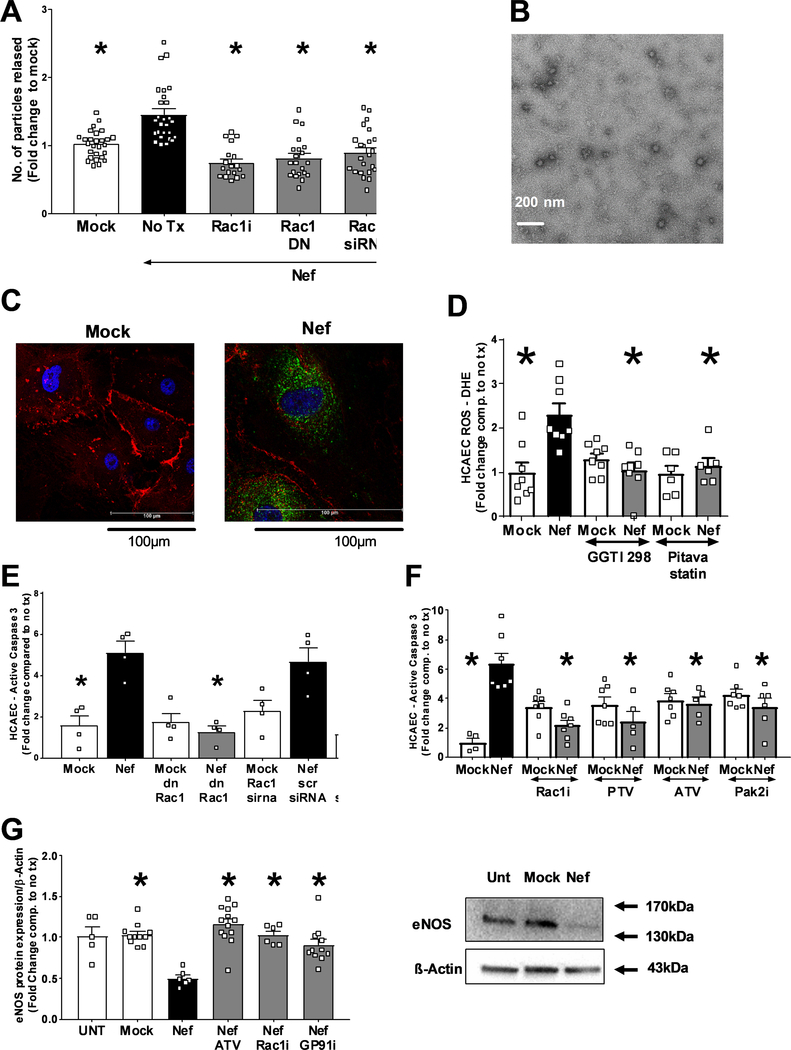
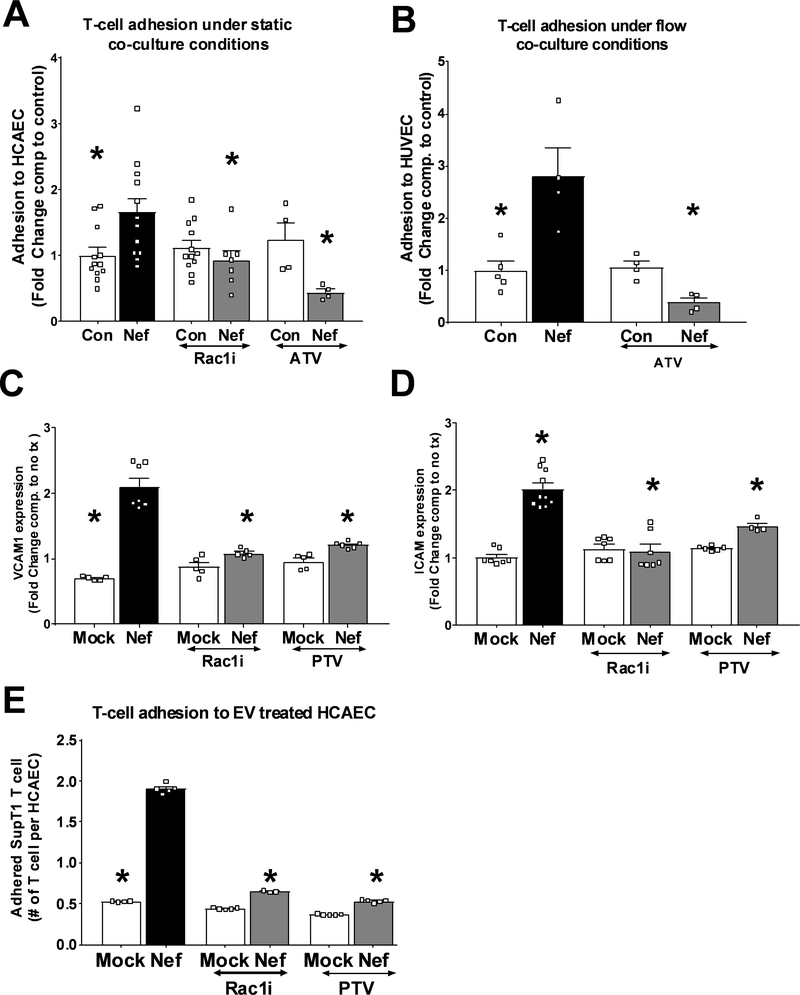
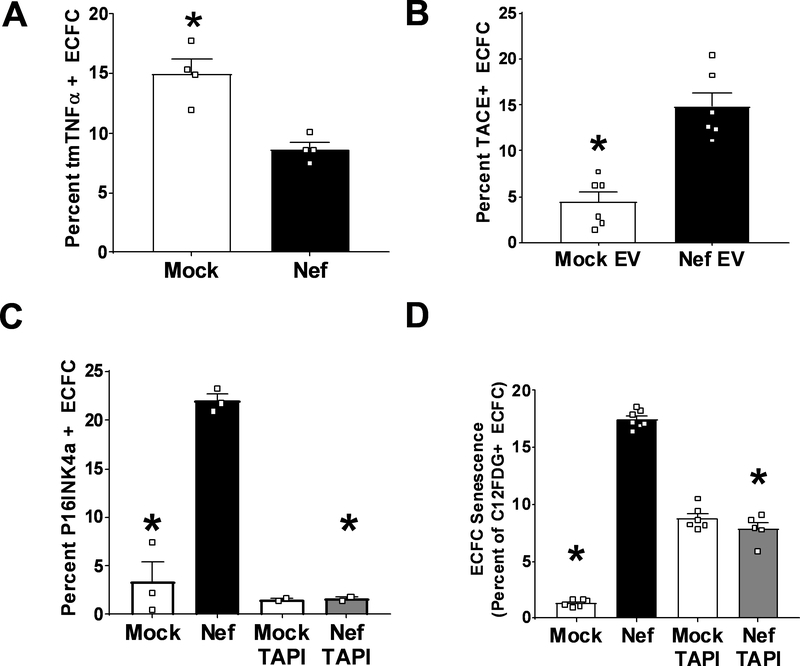
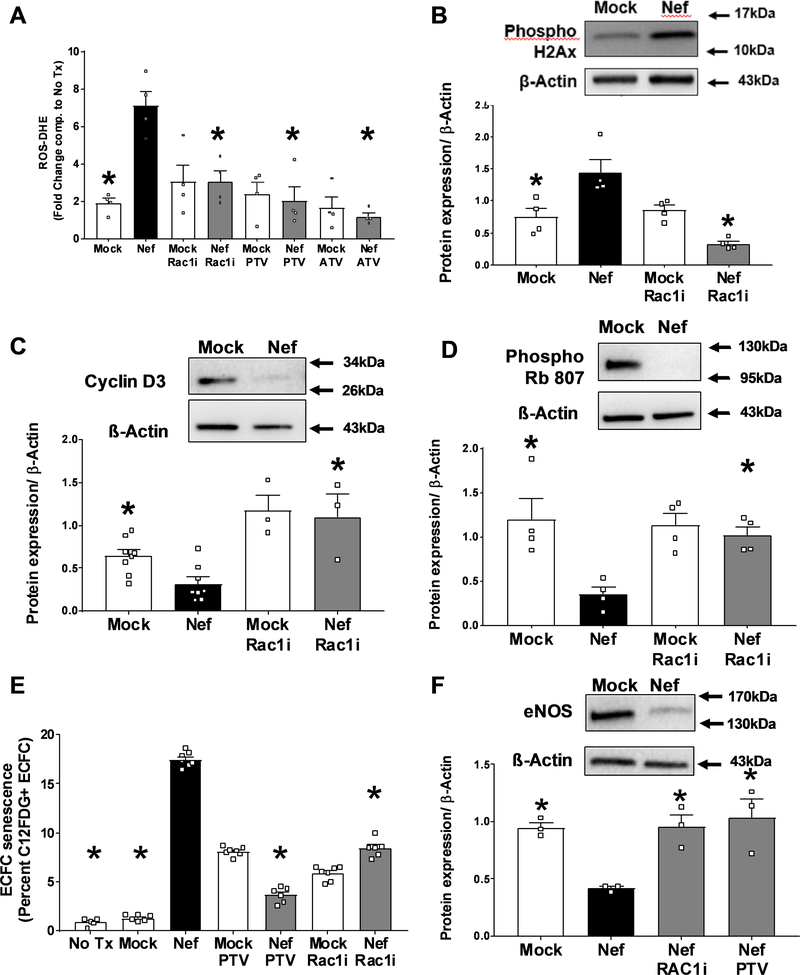
Extracellular vesicles derived from both peripheral blood mononuclear cells and plasma from HIV+ patient blood samples induced human coronary artery endothelial cells dysfunction. Plasma-derived extracellular vesicles from antiretroviral therapy+ patients who were HIV-Nef+ induced significantly greater endothelial apoptosis compared with HIV-Nef-plasma extracellular vesicles. Both HIV-Nef expressing T cells and HIV-Nef-induced extracellular vesicles increased transfer of cytosol and Nef protein to endothelial monolayers in a Rac1-dependent manner, consequently leading to endothelial adhesion protein upregulation and apoptosis. HIV-Nef induced Rac1 activation also led to dsDNA breaks in endothelial colony forming cells, thereby resulting in endothelial colony forming cell premature senescence and endothelial nitric oxide synthase downregulation. These Rac1-dependent activities were characterized by NOX2-mediated reactive oxygen species production. Statin treatment equally inhibited Rac1 inhibition in preventing or reversing all HIV-Nef-induction abnormalities assessed. This was likely because of the ability of statins to block Rac1 prenylation as geranylgeranyl transferase inhibitors were effective in inhibiting HIV-Nef-induced reactive oxygen species formation. Finally, transgenic expression of HIV-Nef in endothelial cells in a murine model impaired endothelium-mediated aortic ring dilation, which was then reversed by 3-week treatment with 5 mg/kg atorvastatin.
These studies establish a mechanism by which HIV-Nef persistence despite antiretroviral therapy could contribute to ongoing HIV-related vascular dysfunction, which may then be ameliorated by statin treatment.
即使在接受抗逆转录病毒治疗的患者中,HIV 仍继续在心血管疾病中发挥致病作用。一个可能的协同因素可能是早期 HIV 反应基因 Nef 的持续存在,我们最近已经证明,在接受抗逆转录病毒治疗的 HIV+患者的肺部中,Nef 持续存在。以前,我们已经报道过,具有 Nef 的 HIV 株,但不是没有 Nef 的 HIV 株,会导致内皮细胞的促炎激活和凋亡。
使用离体组织培养方法以及转基因小鼠模型中的干预实验,描述 HIV-Nef 导致心血管疾病发展的机制。
来自 HIV+患者血液样本的外周血单核细胞和血浆衍生的细胞外囊泡导致人冠状动脉内皮细胞功能障碍。来自接受抗逆转录病毒治疗且 HIV-Nef+的患者的血浆衍生的细胞外囊泡诱导的内皮细胞凋亡明显大于 HIV-Nef-血浆细胞外囊泡。表达 HIV-Nef 的 T 细胞和 HIV-Nef 诱导的细胞外囊泡均以 Rac1 依赖性方式增加细胞质和 Nef 蛋白向内皮单层的转移,从而导致内皮粘附蛋白的上调和凋亡。HIV-Nef 诱导的 Rac1 激活也导致内皮集落形成细胞中的双链 DNA 断裂,从而导致内皮集落形成细胞过早衰老和内皮型一氧化氮合酶下调。这些 Rac1 依赖性活性的特征是由 NOX2 介导的活性氧物质产生。他汀类药物治疗同样通过抑制 Rac1 抑制来预防或逆转评估的所有 HIV-Nef 诱导的异常,这可能是因为他汀类药物能够阻断 Rac1 的异戊烯化,因为 geranylgeranyl 转移酶抑制剂可以有效抑制 HIV-Nef 诱导的活性氧物质形成。最后,在一种小鼠模型中,HIV-Nef 在血管内皮细胞中的转基因表达损害了内皮介导的主动脉环扩张,而经过 3 周的 5mg/kg 阿托伐他汀治疗后,这种损害得到了逆转。
这些研究确立了一种机制,即尽管接受抗逆转录病毒治疗,HIV-Nef 的持续存在仍可能导致持续的 HIV 相关血管功能障碍,而他汀类药物治疗可能会改善这种情况。