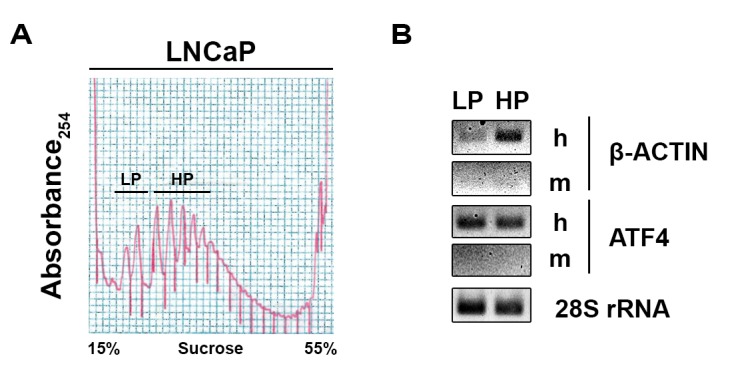
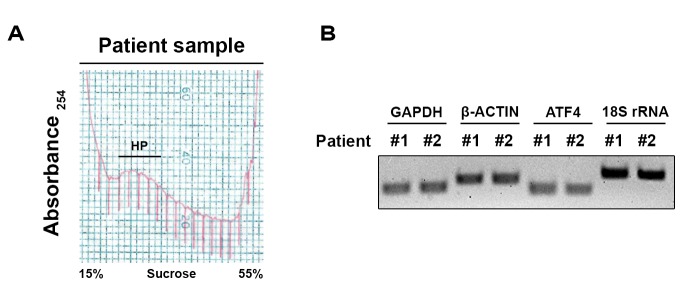
Adjibade Pauline, Grenier St-Sauveur Valérie, Droit Arnaud, Khandjian Edouard W, Toren Paul, Mazroui Rachid
Centre de Recherche en Cancérologie. Centre de Recherche du CHU de Québec. Département de Biologie Moléculaire, Biochimie Médicale et Pathologie, Faculté de Médecine, Université Laval, Québec, PQ, Canada.
Centre de Recherche du CHU de Québec. Département de Médecine Moléculaire, Faculté de Médecine, Université Laval, Québec, PQ, Canada.
J Biol Methods. 2016 Nov 21;3(4):e59. doi: 10.14440/jbm.2016.151. eCollection 2016.
Gene expression involves multiple steps from the transcription of a mRNA in the nucleus to the production of the encoded protein in the cytoplasm. This final step occurs through a highly regulated process of mRNA translation on ribosomes that is required to maintain cell homeostasis. Alterations in the control of mRNA translation may lead to cell's transformation, a hallmark of cancer development. Indeed, recent advances indicated that increased translation of mRNAs encoding tumor-promoting proteins may be a key mechanism of tumor resistance in several cancers. Moreover, it was found that proteins whose encoding mRNAs are translated at higher efficiencies may be effective biomarkers. Evaluation of global changes in translation efficiency in human tumors has thus the potential of better understanding what can be used as biomarkers and therapeutic targets. Investigating changes in translation efficiency in human cancer cells has been made possible through the development and use of the polyribosome profiling combined with DNA microarray or deep RNA sequencing (RNA-Seq). While helpful, the use of cancer cell lines has many limitations and it is essential to define translational changes in human tumor samples in order to properly prioritize genes implicated in cancer phenotype. We present an optimized polyribosome RNA-Seq protocol suitable for quantitative analysis of mRNA translation that occurs in human tumor samples and murine xenografts. Applying this innovative approach to human tumors, which requires a complementary bioinformatics analysis, unlocks the potential to identify key mRNA which are preferentially translated in tumor tissue compared to benign tissue as well as translational changes which occur following treatment. These technical advances will be of interest to those researching all solid tumors, opening possibilities for understanding what may be therapeutic Achilles heels' or relevant biomarkers.
基因表达涉及多个步骤,从细胞核中mRNA的转录到细胞质中编码蛋白质的产生。这最后一步通过核糖体上高度调控的mRNA翻译过程发生,该过程对于维持细胞内稳态是必需的。mRNA翻译控制的改变可能导致细胞转化,这是癌症发展的一个标志。事实上,最近的进展表明,编码促肿瘤蛋白的mRNA翻译增加可能是几种癌症中肿瘤耐药的关键机制。此外,发现其编码mRNA以更高效率翻译的蛋白质可能是有效的生物标志物。因此,评估人类肿瘤中翻译效率的整体变化有可能更好地理解哪些可以用作生物标志物和治疗靶点。通过开发和使用结合DNA微阵列或深度RNA测序(RNA-Seq)的多核糖体谱分析,研究人类癌细胞中翻译效率的变化成为可能。虽然有帮助,但使用癌细胞系有许多局限性,为了正确确定与癌症表型相关的基因优先级,定义人类肿瘤样本中的翻译变化至关重要。我们提出了一种优化的多核糖体RNA-Seq方案,适用于对人类肿瘤样本和小鼠异种移植中发生的mRNA翻译进行定量分析。将这种创新方法应用于人类肿瘤,这需要互补的生物信息学分析,释放了识别与良性组织相比在肿瘤组织中优先翻译的关键mRNA以及治疗后发生的翻译变化的潜力。这些技术进展将引起所有实体瘤研究人员的兴趣,为理解什么可能是治疗的“阿喀琉斯之踵”或相关生物标志物开辟了可能性。