Li Xue, Wei Donghui, Li Zhongjun
The College of Chemistry and Molecular Engineering, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan 450001, P. R. China.
ACS Omega. 2017 Oct 20;2(10):7029-7038. doi: 10.1021/acsomega.7b00907. eCollection 2017 Oct 31.
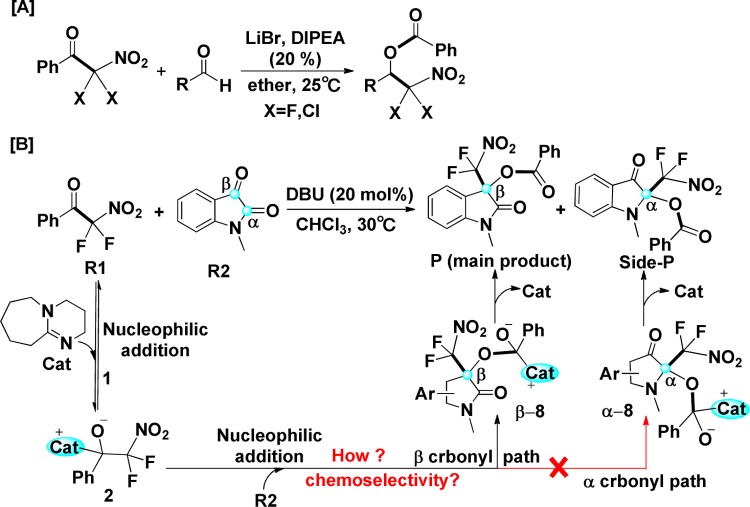
The possible mechanisms of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)-catalyzed chemoselective insertion of -methyl isatin into aryl difluoronitromethyl ketone to synthesize 3,3-disubstituted and 2,2-disubstituted oxindoles have been studied in this work. As revealed by calculated results, the reaction occurs via two competing paths, including α and β carbonyl paths, and each path contains five steps, that is, nucleophilic addition of DBU to ketone, C-C bond cleavage affording difluoromethylnitrate anion and phenylcarbonyl-DBU cation, nucleophilic addition of difluoromethylnitrate anion to carbonyl carbon of -methyl isatin, acyl transfer process, and dissociation of DBU and product. The computational results suggest that nucleophilic additions on different carbonyl carbons of -methyl isatin via α and β carbonyl paths would lead to different products in the third step, and β carbonyl path associated with the main product 3,3-disubstituted oxindole is more energetically favorable, which is consistent with the experimental observations. Noteworthy, electrophilic Parr function can be successfully applied for exactly predicting the activity of reaction site and reasonably explaining the chemoselectivity. In addition, the distortion/interaction and noncovalent interaction analyses show that much more hydrogen bond interactions should be responsible for the lower energy of the transition state associated with β carbonyl path. The obtained insights would be valuable for the rational design of efficient organocatalysts for this kind of reactions with high selectivities.
本工作研究了1,8 - 二氮杂双环[5.4.0]十一碳 - 7 - 烯(DBU)催化的α - 甲基异吲哚与芳基二氟硝基甲基酮的化学选择性插入反应,以合成3,3 - 二取代和2,2 - 二取代的氧化吲哚。计算结果表明,该反应通过两条竞争路径发生,包括α和β羰基路径,每条路径包含五个步骤,即DBU对酮的亲核加成、C - C键断裂生成二氟甲基硝酸根阴离子和苯基羰基 - DBU阳离子、二氟甲基硝酸根阴离子对α - 甲基异吲哚羰基碳的亲核加成、酰基转移过程以及DBU和产物的解离。计算结果表明,通过α和β羰基路径在α - 甲基异吲哚的不同羰基碳上进行亲核加成会在第三步产生不同的产物,并且与主要产物3,3 - 二取代氧化吲哚相关的β羰基路径在能量上更有利,这与实验观察结果一致。值得注意的是,亲电帕尔函数可以成功地用于准确预测反应位点的活性并合理解释化学选择性。此外,畸变/相互作用和非共价相互作用分析表明,更多的氢键相互作用是β羰基路径相关过渡态能量较低的原因。所获得的见解对于合理设计用于此类具有高选择性反应的高效有机催化剂具有重要价值。