Gao Peng, Wang Xingyong, Huang Zhenguo, Yu Haibo
School of Chemistry and Molecular Bioscience and Molecular Horizons, University of Wollongong, Wollongong, New South Wales 2500, Australia.
School of Civil and Environmental Engineering, University of Technology Sydney, Sydney, New South Wales 2007, Australia.
ACS Omega. 2019 Jul 19;4(7):12385-12392. doi: 10.1021/acsomega.9b01566. eCollection 2019 Jul 31.
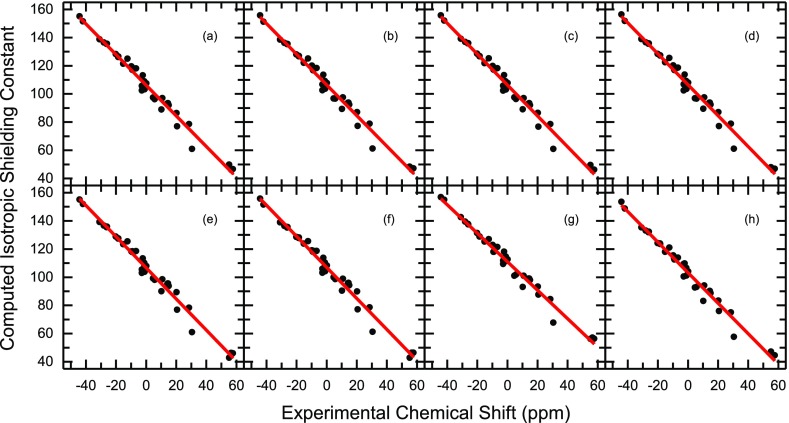
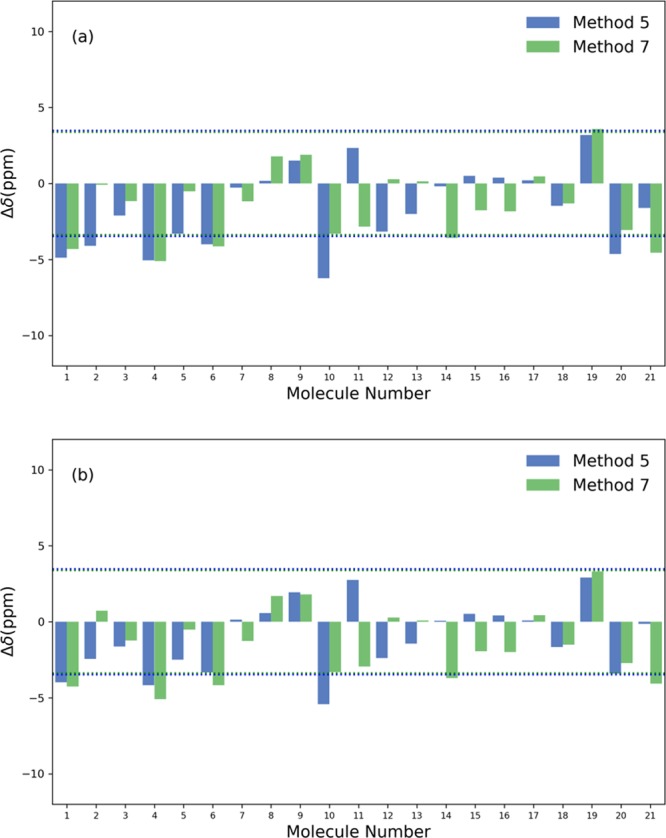
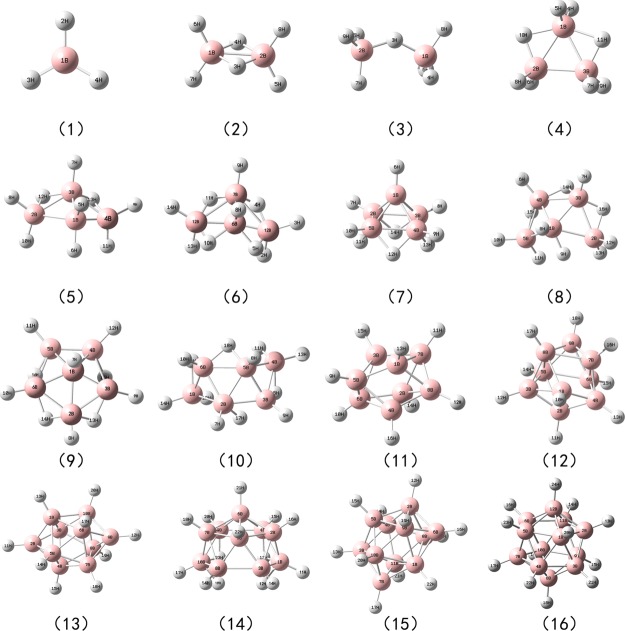
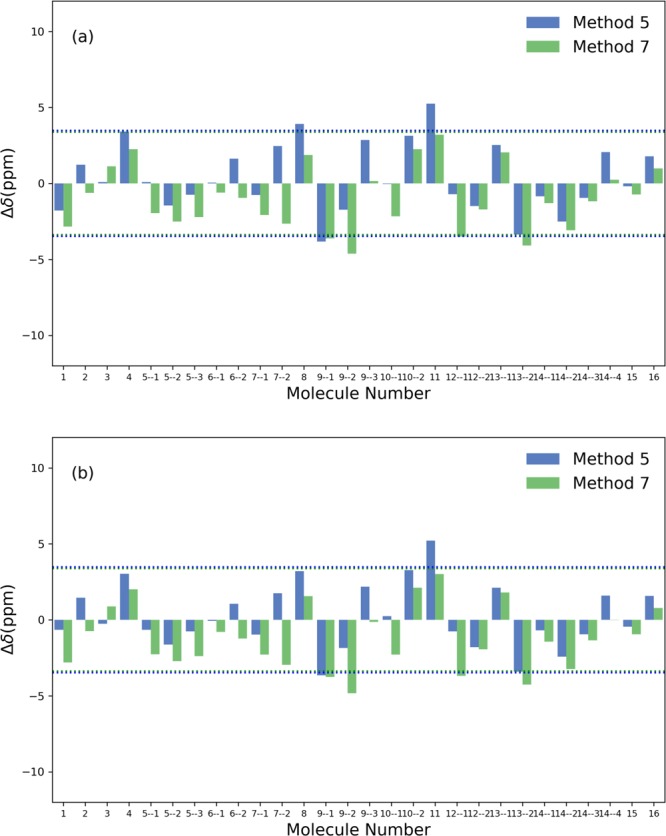
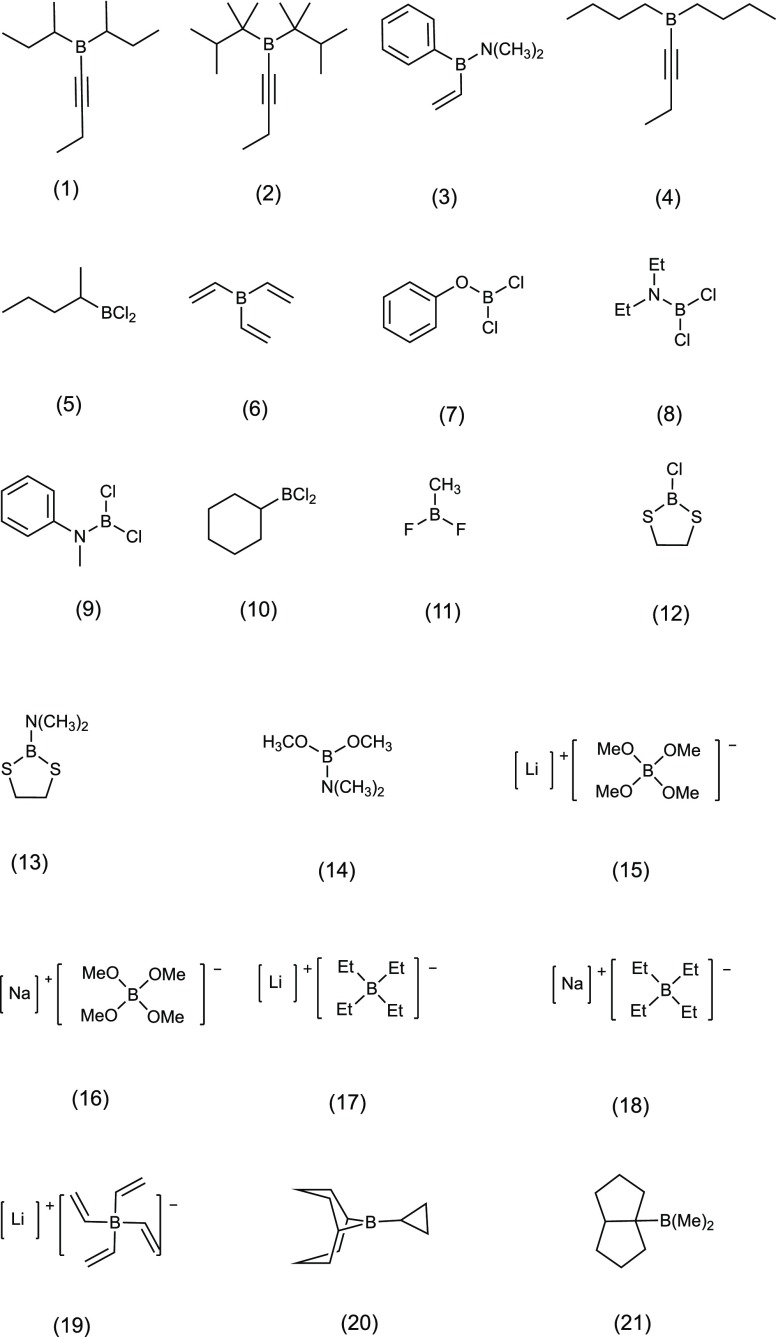
B nuclear magnetic resonance (NMR) spectroscopy is a useful tool for studies of boron-containing compounds in terms of structural analysis and reaction kinetics monitoring. A computational protocol, which is aimed at an accurate prediction of B NMR chemical shifts via linear regression, was proposed based on the density functional theory and the gauge-including atomic orbital approach. Similar to the procedure used for carbon, hydrogen, and nitrogen chemical shift predictions, a database of boron-containing molecules was first compiled. Scaling factors for the linear regression between calculated isotropic shielding constants and experimental chemical shifts were then fitted using eight different levels of theory with both the solvation model based on density and conductor-like polarizable continuum model solvent models. The best method with the two solvent models yields a root-mean-square deviation of about 3.40 and 3.37 ppm, respectively. To explore the capabilities and potential limitations of the developed protocols, classical boron-hydrogen compounds and molecules with representative boron bonding environments were chosen as test cases, and the consistency between experimental values and theoretical predictions was demonstrated.
硼核磁共振(NMR)光谱学是研究含硼化合物结构分析和反应动力学监测的有用工具。基于密度泛函理论和含规范原子轨道方法,提出了一种通过线性回归准确预测硼核磁共振化学位移的计算方法。与用于碳、氢和氮化学位移预测的过程类似,首先编制了一个含硼分子数据库。然后使用基于密度的溶剂化模型和类导体极化连续介质模型这两种溶剂模型,在八个不同的理论水平下拟合计算的各向同性屏蔽常数与实验化学位移之间线性回归的比例因子。两种溶剂模型的最佳方法分别产生约3.40和3.37 ppm的均方根偏差。为了探索所开发方法的能力和潜在局限性,选择了经典的硼氢化合物和具有代表性硼键环境的分子作为测试案例,并证明了实验值与理论预测之间的一致性。