Nonaka Kentaro, Han Xu, Kato Hiroki, Sato Hiroshi, Yamaza Haruyoshi, Hirofuji Yuta, Masuda Keiji
Department of Surgery and Science, Graduate School of Medical Sciences, Kyushu University, Maidashi 3-1-1, Higashi-Ku, Fukuoka, 812-8582, Japan.
Section of Oral Medicine for Children, Division of Oral Health, Growth and Development, Faculty of Dental Science, Kyushu University, Maidashi 3-1-1, Higashi-Ku, Fukuoka, 812-8582, Japan.
Biochem Biophys Rep. 2019 May 17;19:100648. doi: 10.1016/j.bbrep.2019.100648. eCollection 2019 Sep.
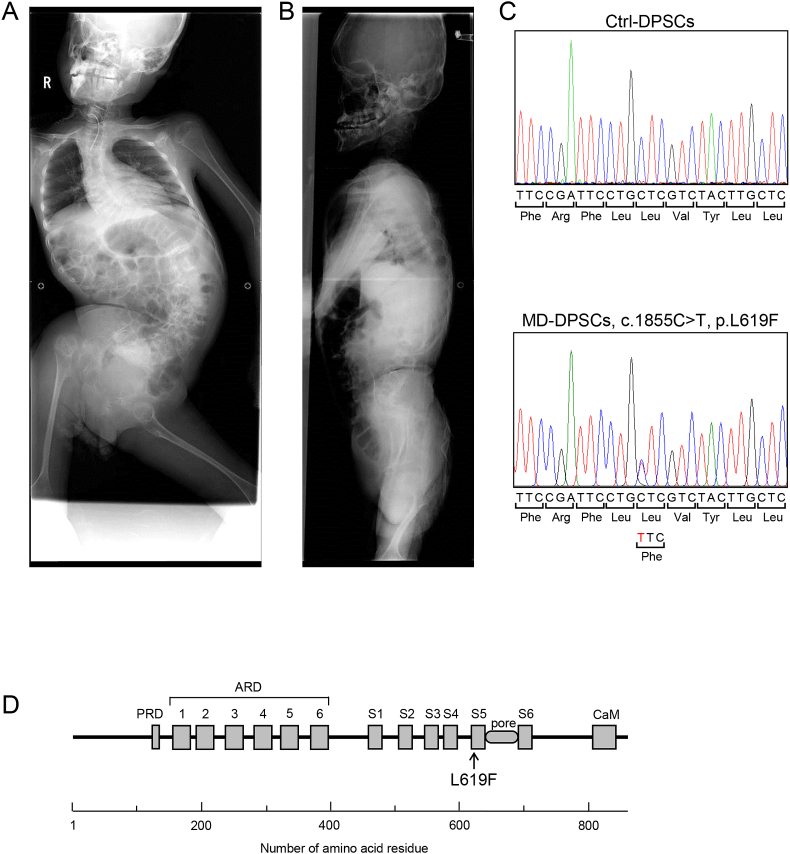
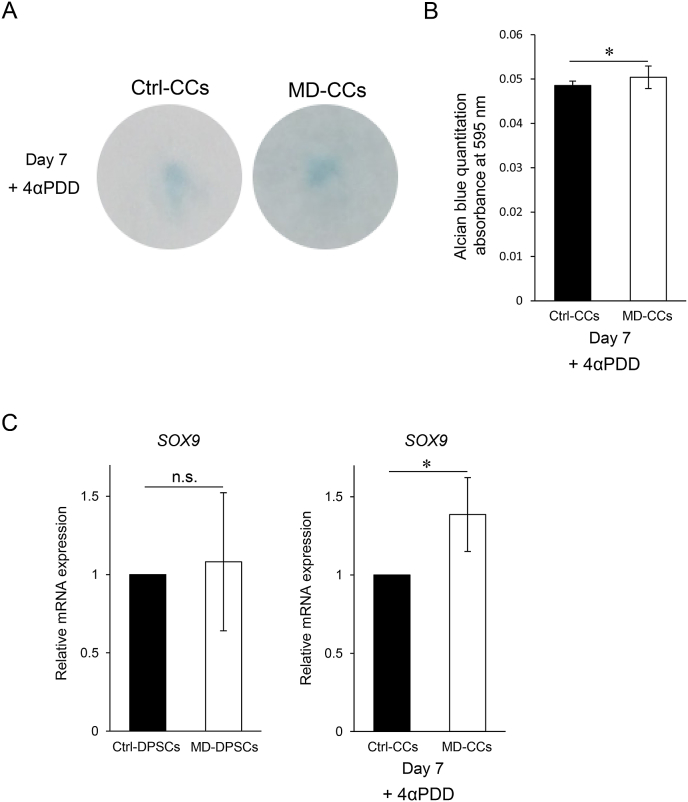
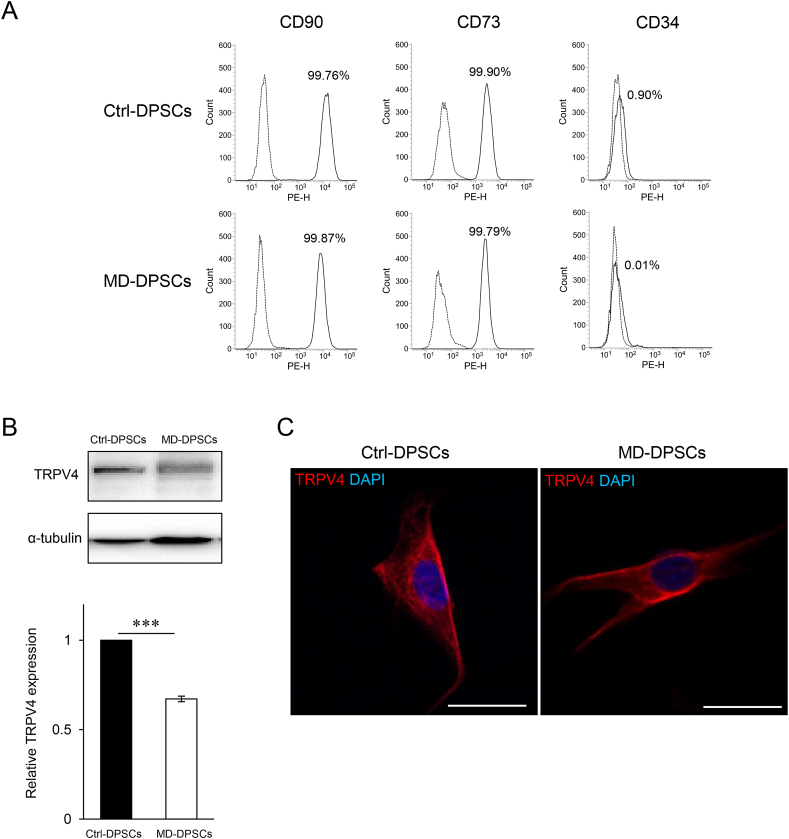
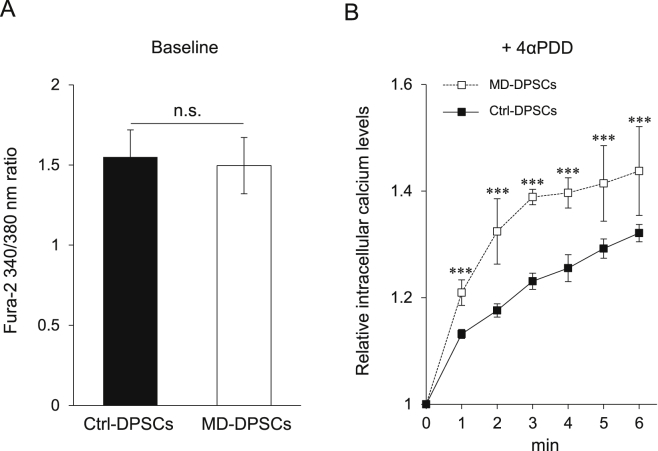
Metatropic dysplasia is a congenital skeletal dysplasia characterized by severe platyspondyly, dumbbell-like deformity of long tubular bones, and progressive kyphoscoliosis with growth. It is caused by mutations in the gene , encoding the transient receptor potential vanilloid 4, which acts as a calcium channel. Many heterozygous single base mutations of this gene have been associated with the disorder, showing autosomal dominant inheritance. Although abnormal endochondral ossification has been observed by histological examination of bone in a patient with lethal metatropic dysplasia, the etiology of the disorder remains largely unresolved. As dental pulp stem cells (DPSCs) are mesenchymal stem cells that differentiate into bone lineage cells, DPSCs derived from patients with congenital skeletal dysplasia might be useful as a disease-specific cellular model for etiological investigation. The purpose of this study was to clarify the pathological association between mutation and chondrocyte differentiation by analyzing DPSCs from a patient with non-lethal metatropic dysplasia. We identified a novel heterozygous single base mutation, c.1855C>T in . This was predicted to be a missense mutation, p.L619F, in putative transmembrane segment 5. The mutation was repaired by CRISPR/Cas9 system to obtain isogenic control DPSCs for further analysis. The expression of stem cell markers and fibroblast-like morphology were comparable between patient-derived mutant and control DPSCs, although expression of TRPV4 was lower in mutant DPSCs than control DPSCs. Despite the lower TRPV4 expression in mutant DPSCs, the intracellular Ca level was comparable at the basal level between mutant and control DPSCs, while its level was markedly higher following stimulation with 4α-phorbol 12,13-didecanoate (4αPDD), a specific agonist for TRPV4, in mutant DPSCs than in control DPSCs. In the presence of 4αPDD, we observed accelerated early chondrocyte differentiation and upregulated mRNA expression of SRY-box 9 () in mutant DPSCs. Our findings suggested that the novel missense mutation c.1855C>T of was a gain-of-function mutation leading to enhanced intracellular Ca level, which was associated with accelerated chondrocyte differentiation and upregulation. Our results also suggest that patient-derived DPSCs can be a useful disease-specific cellular model for elucidating the pathological mechanism of metatropic dysplasia.
变异性发育不良是一种先天性骨骼发育不良,其特征为严重的扁平椎、长管状骨的哑铃状畸形以及随生长而进展的脊柱侧凸。它由编码瞬时受体电位香草酸受体4(TRPV4)的基因突变引起,TRPV4作为一种钙通道发挥作用。该基因的许多杂合单碱基突变与这种疾病相关,呈常染色体显性遗传。尽管通过对一名致死性变异性发育不良患者的骨组织学检查观察到软骨内成骨异常,但该疾病的病因在很大程度上仍未得到解决。由于牙髓干细胞(DPSCs)是可分化为骨谱系细胞的间充质干细胞,来自先天性骨骼发育不良患者的DPSCs可能作为病因学研究的疾病特异性细胞模型。本研究的目的是通过分析一名非致死性变异性发育不良患者的DPSCs来阐明TRPV4突变与软骨细胞分化之间的病理关联。我们在TRPV4中鉴定出一个新的杂合单碱基突变,c.1855C>T。这被预测为一个错义突变,p.L619F,位于假定的跨膜区段5。通过CRISPR/Cas9系统修复该突变以获得等基因对照DPSCs用于进一步分析。患者来源的突变型和对照DPSCs之间干细胞标志物的表达和成纤维细胞样形态相当,尽管突变型DPSCs中TRPV4的表达低于对照DPSCs。尽管突变型DPSCs中TRPV4表达较低,但突变型和对照DPSCs在基础水平时细胞内钙水平相当,而在用TRPV4特异性激动剂4α-佛波醇12,13-十四烷酸酯(4αPDD)刺激后,突变型DPSCs中的钙水平明显高于对照DPSCs。在存在4αPDD的情况下,我们观察到突变型DPSCs中早期软骨细胞分化加速且SRY盒9(SOX9)的mRNA表达上调。我们的研究结果表明,TRPV4的新错义突变c.1855C>T是一个功能获得性突变,导致细胞内钙水平升高,这与软骨细胞分化加速和SOX /span>上调相关。我们的结果还表明,患者来源的DPSCs可以作为阐明变异性发育不良病理机制的有用的疾病特异性细胞模型。