National Institutes of Health, NINDS, NNDCS, Bethesda, Maryland.
Department of Neurology, Mayo Clinic, Rochester, Minnesota.
Ann Clin Transl Neurol. 2019 Oct;6(10):1980-1988. doi: 10.1002/acn3.50882. Epub 2019 Sep 11.
To characterize the natural history and clinical features of myopathies caused by mono-allelic, dominantly acting pathogenic variants in COL12A1.
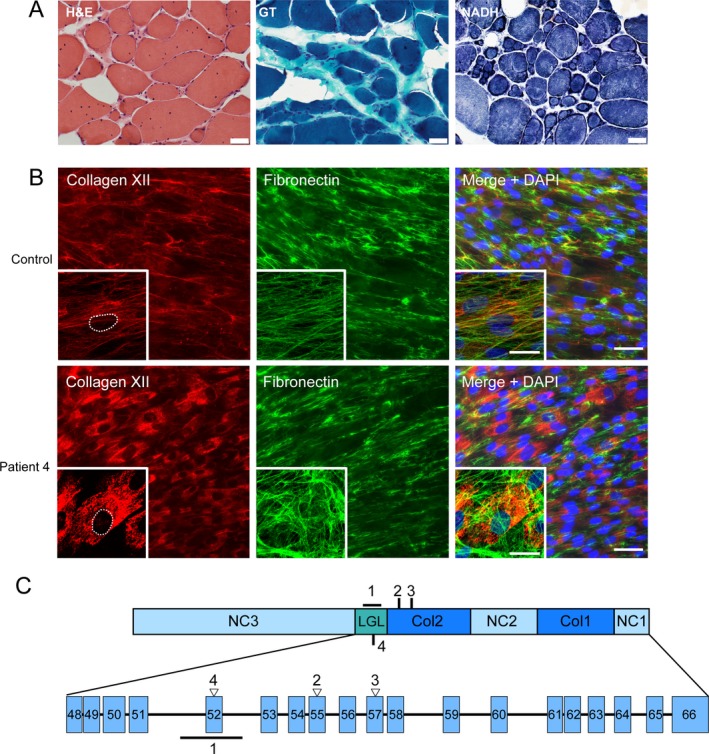
Patients with dominant COL12A1-related myopathies were characterized by history and clinical examination, muscle imaging, and genetic analysis. Pathogenicity of the variants was assessed by immunostaining patient-derived dermal fibroblast cultures for collagen XII.
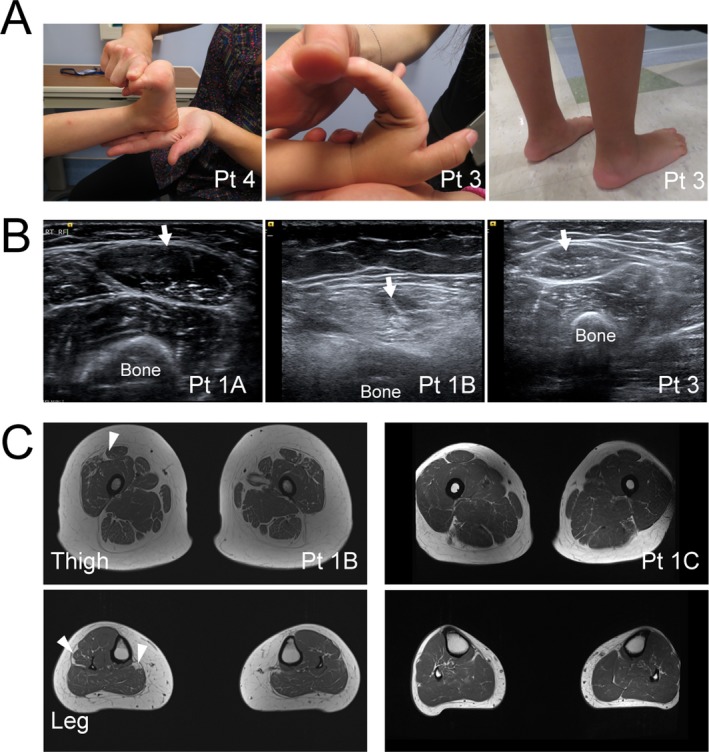
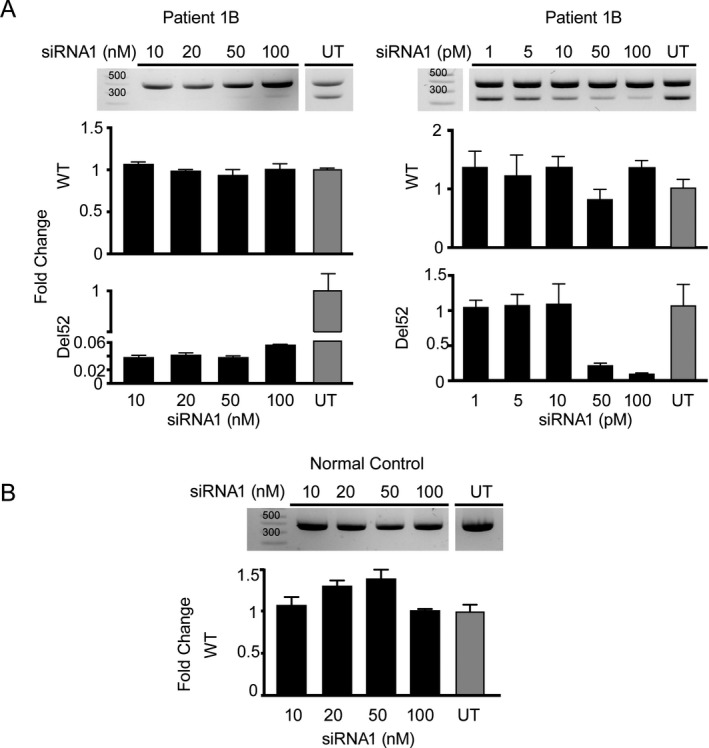
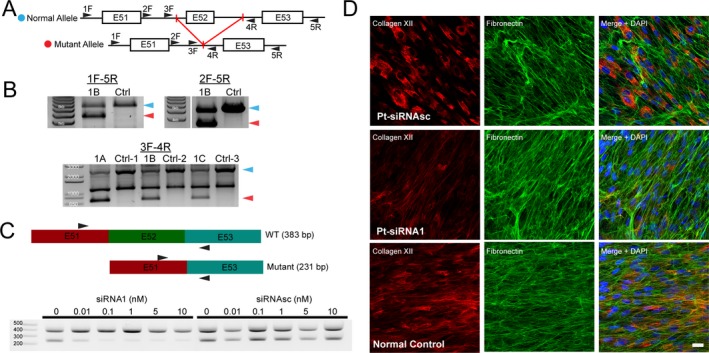
Four independent families with childhood-onset weakness due to novel, dominantly acting pathogenic variants in COL12A1 were identified. Adult patients exhibited distal-predominant weakness. Three families carried dominantly acting glycine missense variants, and one family had a heterozygous, intragenic, in-frame deletion of exon 52 of COL12A1. All pathogenic variants resulted in increased intracellular retention of collagen XII in patient-derived fibroblasts as well as loss of extracellular, fibrillar collagen XII deposition. Since haploinsufficiency for COL12A1 is largely clinically asymptomatic, we designed and evaluated small interfering RNAs (siRNAs) that specifically target the mutant allele containing the exon 52 deletion. Immunostaining of the patient fibroblasts treated with the siRNA showed a near complete correction of collagen XII staining patterns.
This study characterizes a distal myopathy phenotype in adults with dominant COL12A1 pathogenic variants, further defining the phenotypic spectrum and natural history of COL12A1-related myopathies. This work also provides proof of concept of a precision medicine treatment approach by proposing and validating allele-specific knockdown using siRNAs specifically designed to target a patient's dominant COL12A1 disease allele.
描述由 COL12A1 单等位基因显性致病变异引起的肌病的自然病史和临床特征。
通过病史和临床检查、肌肉影像学和基因分析对具有显性 COL12A1 相关肌病的患者进行特征描述。通过免疫染色患者来源的皮肤成纤维细胞中的胶原 XII 来评估变体的致病性。
确定了四个具有 COL12A1 新的显性致病变异的独立家族,这些家族的患者均患有儿童期起病的肌无力。成年患者表现出远端为主的肌无力。三个家族携带显性作用的甘氨酸错义变异,一个家族携带 COL12A1 外显子 52 的杂合、内含子内、框内缺失。所有致病变异均导致 COL12A1 患者来源的成纤维细胞内胶原 XII 的细胞内滞留增加,以及细胞外纤维状胶原 XII 沉积丢失。由于 COL12A1 的杂合不足在很大程度上无临床症状,我们设计并评估了专门针对包含外显子 52 缺失的突变等位基因的小干扰 RNA (siRNA)。用 siRNA 处理的患者成纤维细胞的免疫染色显示胶原 XII 染色模式几乎完全纠正。
本研究描述了具有显性 COL12A1 致病变异的成年人的远端肌病表型,进一步定义了 COL12A1 相关肌病的表型谱和自然病史。这项工作还通过提出并验证了专门针对患者显性 COL12A1 疾病等位基因的等位基因特异性敲低的 siRNA,为精准医学治疗方法提供了概念验证。