McCarty Riley M, Saade Dimah, Munot Pinki, Laverty Chamindra G, Pinz Hailey, Zou Yaqun, McAnally Meghan, Yun Pomi, Tian Cuixia, Hu Ying, Feng Lucy, Phadke Rahul, Ceulemans Sophia, Magoulas Pilar, Skalsky Andrew J, Friedman Jennifer R, Braddock Stephen R, Neuhaus Sarah B, Malicki Denise M, Bainbridge Matthew N, Nahas Shareef, Dimmock David P, Kingsmore Stephen F, Lotze Timothy E, Foley A Reghan, Muntoni Francesco, Straub Volker, Donkervoort Sandra, Bönnemann Carsten G
Neuromuscular and Neurogenetic Disorders of Childhood Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA.
Department of Pediatrics, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Ann Clin Transl Neurol. 2025 Mar;12(3):602-614. doi: 10.1002/acn3.52225. Epub 2025 Feb 9.
While there have been several reports of patients with dominantly acting COL12A1 variants, few cases of the more severe recessive Collagen XII-related disorders have previously been documented.
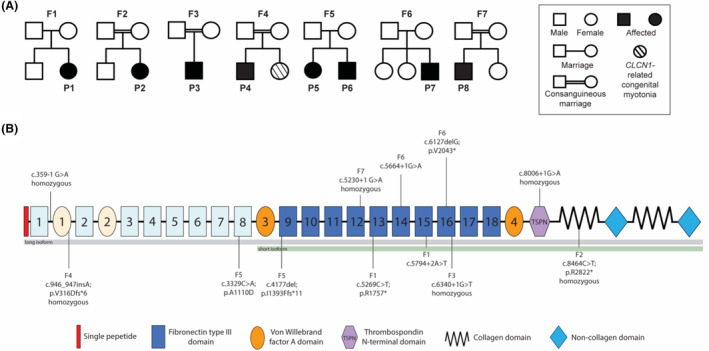
We present detailed clinical, immunocytochemical, and imaging data on eight additional patients from seven families with biallelic pathogenic variants in COL12A1.
All patients presented with a consistent constellation of congenital onset clinical features: hypotonia, dysmorphic features, most notably gingival hypertrophy, prominent distal joint hyperlaxity, with co-occurring contractures of large joints, and variable muscle involvement, evident both clinically and on muscle imaging. Five patients presented with a severe congenital phenotype manifesting with profound weakness, significantly delayed or minimal attainment of motor milestones, respiratory insufficiency, and feeding difficulties. Three patients presented with mild-to-moderate muscle weakness and delayed milestones but were able to achieve independent ambulation. Patients were found to have biallelic loss-of-function COL12A1 variants, except for one family (p.I1393Ffs*11/p.A1110D). Consistent with the variable clinical spectrum, in vitro immunocytochemistry analysis in fibroblasts ranged from complete absence of Collagen XII expression in a patient with severe disease, to a mild reduction in a patient with milder disease.
Here we characterize the clinical presentation, muscle imaging, and dermal fibroblast immunostaining findings associated with biallelic variants in COL12A1, further establishing COL12A1 as a recessive myopathic Ehlers-Danlos syndrome (mEDS) gene, and expanding the clinical spectrum to include a milder EDS phenotype.
虽然已有多篇关于显性作用的COL12A1变异患者的报道,但此前记录的更为严重的隐性胶原XII相关疾病病例较少。
我们展示了来自7个家庭的另外8例患者的详细临床、免疫细胞化学和影像学数据,这些患者的COL12A1存在双等位基因致病变异。
所有患者均表现出一系列一致的先天性临床特征:肌张力减退、畸形特征,最显著的是牙龈增生、明显的远端关节过度松弛,同时伴有大关节挛缩,以及不同程度的肌肉受累,在临床和肌肉影像学上均有体现。5例患者表现出严重的先天性表型,表现为严重肌无力、运动里程碑显著延迟或进展极小、呼吸功能不全和喂养困难。3例患者表现为轻度至中度肌无力和发育里程碑延迟,但能够独立行走。除一个家系(p.I1393Ffs*11/p.A1110D)外,患者均被发现存在双等位基因功能丧失的COL12A1变异。与临床谱的多样性一致,成纤维细胞的体外免疫细胞化学分析显示,严重疾病患者完全没有胶原XII表达,而病情较轻的患者则有轻度减少。
在此,我们描述了与COL12A1双等位基因变异相关的临床表现、肌肉影像学和皮肤成纤维细胞免疫染色结果,进一步确定COL12A1为隐性肌病性埃勒斯-当洛综合征(mEDS)基因,并扩大了临床谱,以纳入较轻的EDS表型。