Department of Pharmacology and Physiology, College of Osteopathic Medicine, Oklahoma State University Center for Health Sciences, Tahlequah, OK, USA.
Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Neurochem Int. 2019 Dec;131:104552. doi: 10.1016/j.neuint.2019.104552. Epub 2019 Sep 20.
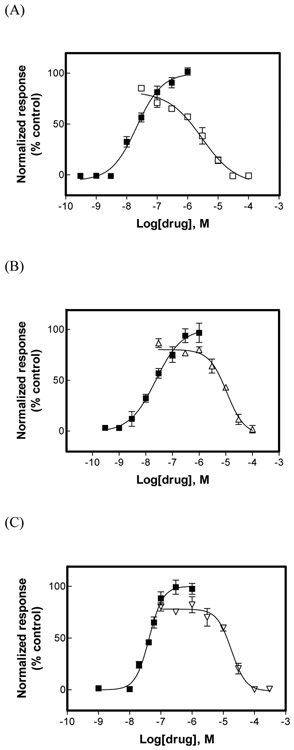
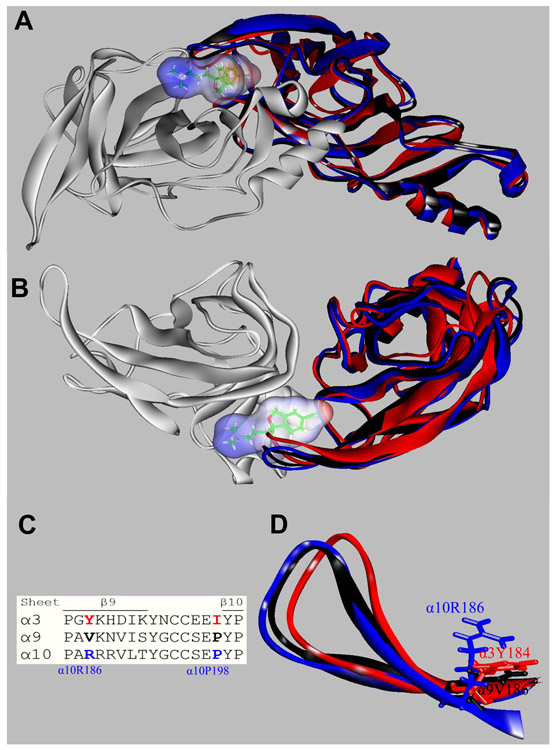
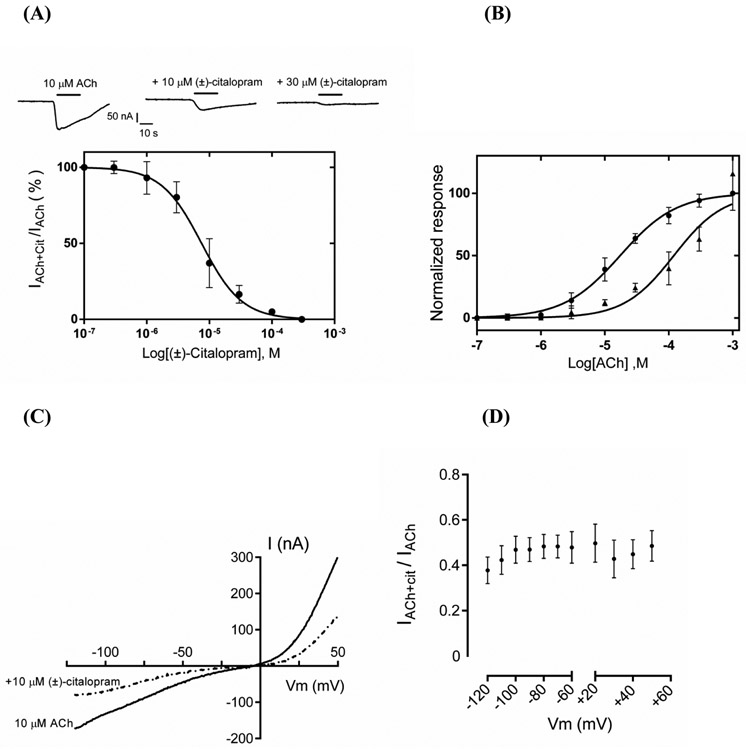
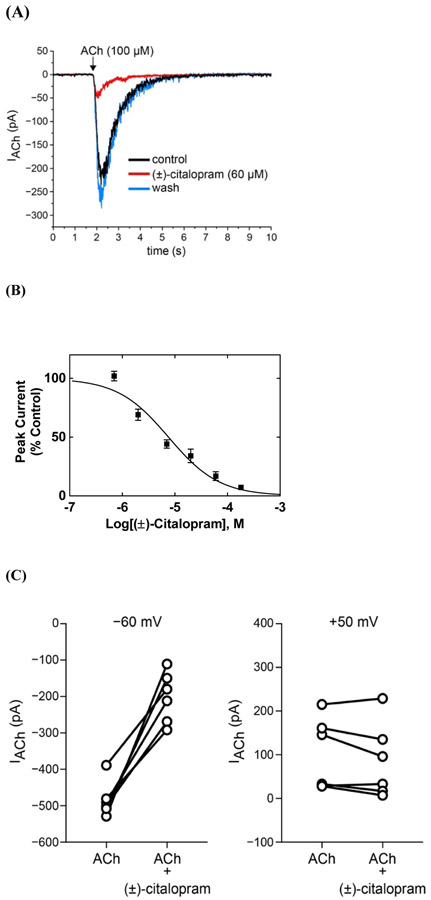
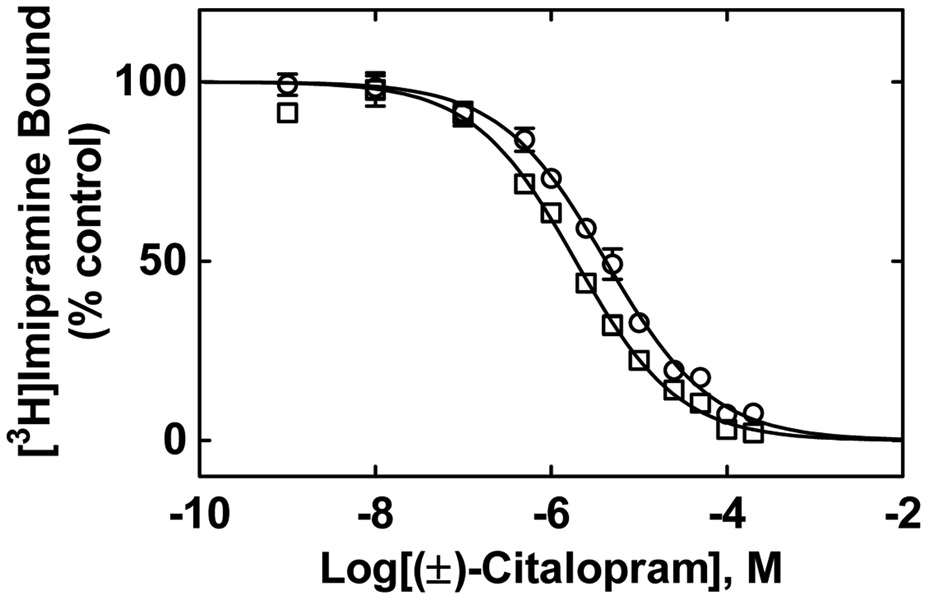
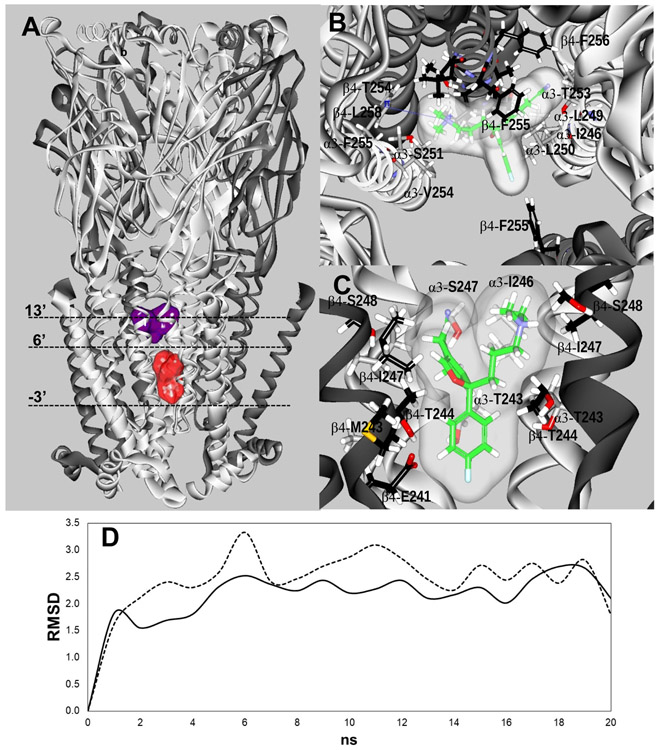
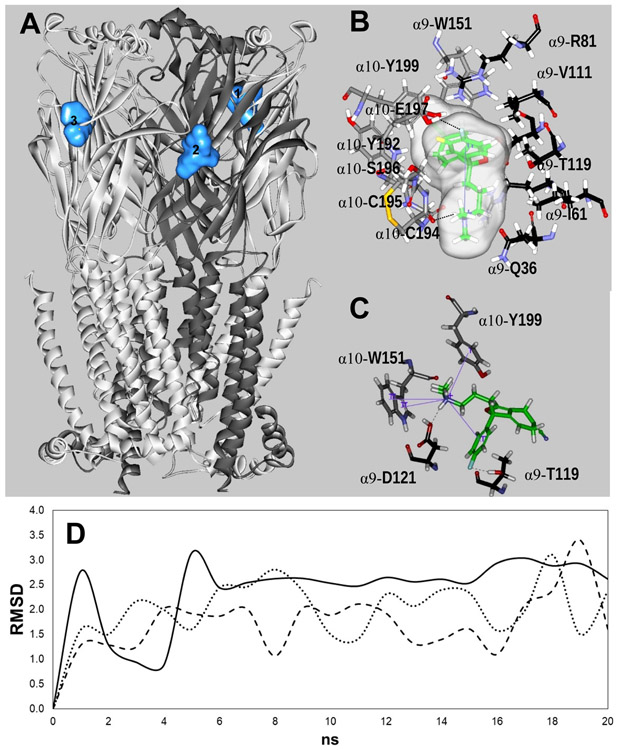
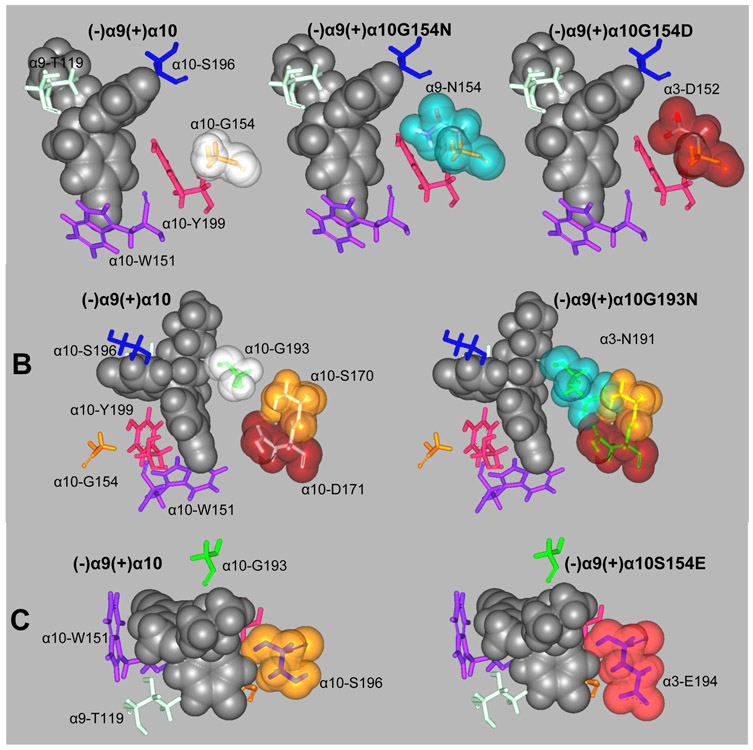
The inhibitory activity of (±)-citalopram on human (h) α3β4, α4β2, and α7 nicotinic acetylcholine receptors (AChRs) was determined by Ca influx assays, whereas its effect on rat α9α10 and mouse habenular α3β4* AChRs by electrophysiological recordings. The Ca influx results clearly establish that (±)-citalopram inhibits (IC's in μM) hα3β4 AChRs (5.1 ± 1.3) with higher potency than that for hα7 (18.8 ± 1.1) and hα4β2 (19.1 ± 4.2) AChRs. This is in agreement with the [H]imipramine competition binding results indicating that (±)-citalopram binds to imipramine sites at desensitized hα3β4 with >2-fold higher affinity than that for hα4β2. The electrophysiological, molecular docking, and in silico mutation results indicate that (±)-citalopram competitively inhibits rα9α10 AChRs (7.5 ± 0.9) in a voltage-independent manner by interacting mainly with orthosteric sites, whereas it inhibits a homogeneous population of α3β4* AChRs at MHb (VI) neurons (7.6 ± 1.0) in a voltage-dependent manner by interacting mainly with a luminal site located in the middle of the ion channel, overlapping the imipramine site, which suggests an ion channel blocking mechanism. In conclusion, (±)-citalopram inhibits α3β4 and α9α10 AChRs with higher potency compared to other AChRs but by different mechanisms. (±)-Citalopram also inhibits habenular α3β4*AChRs, supporting the notion that these receptors are important endogenous targets related to their anti-addictive activities.
(±)-西酞普兰对人(h)α3β4、α4β2 和α7 烟碱型乙酰胆碱受体(AChRs)的抑制活性通过 Ca2+内流测定法确定,而其对大鼠α9α10 和小鼠缰核α3β4AChRs 的影响则通过电生理记录来确定。Ca2+内流结果清楚地表明,(±)-西酞普兰抑制(IC50 以 μM 计)hα3β4 AChRs(5.1±1.3)的活性比 hα7(18.8±1.1)和 hα4β2(19.1±4.2)AChRs 更高。这与[H]丙咪嗪竞争结合结果一致,表明(±)-西酞普兰与脱敏的 hα3β4 上的丙咪嗪结合位点结合的亲和力比 hα4β2 高>2 倍。电生理、分子对接和计算机模拟突变结果表明,(±)-西酞普兰以非电压依赖性方式竞争性抑制 rα9α10 AChRs(7.5±0.9),主要通过与正位位点相互作用,而以电压依赖性方式抑制 MHb(VI)神经元中的同质α3β4AChRs 群体(7.6±1.0),主要通过与位于离子通道中部的腔内位点相互作用,该位点与丙咪嗪位点重叠,提示离子通道阻断机制。总之,(±)-西酞普兰对α3β4 和α9α10 AChRs 的抑制活性比其他 AChRs 更高,但作用机制不同。(±)-西酞普兰还抑制缰核α3β4*AChRs,支持这些受体是与其抗成瘾活性相关的重要内源性靶标的观点。