Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92092-0378.
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92092-0378.
Mol Cell Proteomics. 2019 Dec;18(12):2516-2523. doi: 10.1074/mcp.TIR119.001731. Epub 2019 Oct 8.
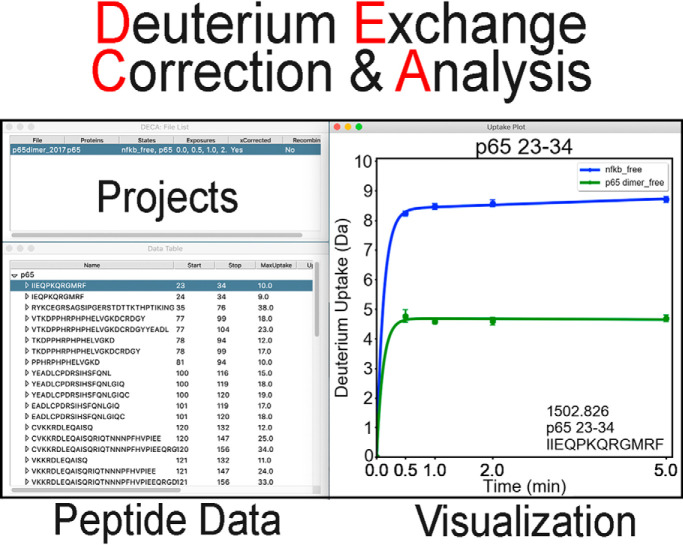
Amide hydrogen-deuterium exchange mass spectrometry (HDX-MS) has become widely popular for mapping protein-ligand interfaces, for understanding protein-protein interactions, and for discovering dynamic allostery. Several platforms are now available which provide large data sets of amide hydrogen/deuterium exchange mass spectrometry (HDX-MS) data. Although many of these platforms provide some down-stream processing, a comprehensive software that provides the most commonly used down-stream processing tools such as automatic back-exchange correction options, analysis of overlapping peptides, calculations of relative deuterium uptake into regions of the protein after such corrections, rigorous statistical analysis of the significance of uptake differences, and generation of high quality figures for data presentation is not yet available. Here we describe the Deuterium Exchange Correction and Analysis (DECA) software package, which provides all these downstream processing options for data from the most popular mass spectrometry platforms. The major functions of the software are demonstrated on sample data.
酰胺氢-氘交换质谱(HDX-MS)已广泛用于绘制蛋白质-配体界面图、了解蛋白质-蛋白质相互作用以及发现动态变构。现在有几个平台可以提供大量的酰胺氢/氘交换质谱(HDX-MS)数据。尽管这些平台中的许多都提供了一些下游处理,但缺乏一个综合性的软件来提供最常用的下游处理工具,如自动回交换校正选项、重叠肽分析、在进行这些校正后计算蛋白质区域内相对氘摄取量、对摄取差异的显著性进行严格的统计分析,以及生成高质量的数据图进行数据展示。在这里,我们描述了氘交换校正和分析(DECA)软件包,它为最流行的质谱平台的数据提供了所有这些下游处理选项。软件的主要功能在示例数据上进行了演示。