Center for Clinical Molecular Medicine, Chongqing, 400014, People's Republic of China.
Ministry of Education Key Laboratory of Child Development and Disorder, Chongqing, 400014, People's Republic of China.
BMC Pediatr. 2019 Oct 9;19(1):344. doi: 10.1186/s12887-019-1747-5.
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase (mHS) deficiency is an autosomal recessive inborn error of metabolism, which will give rise to failure of ketogenesis in liver during illness or fasting. It is a very rare disease with only a few patients reported worldwide, most of which had a good prognosis after proper therapies.
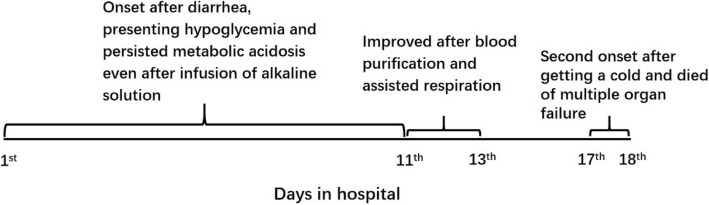
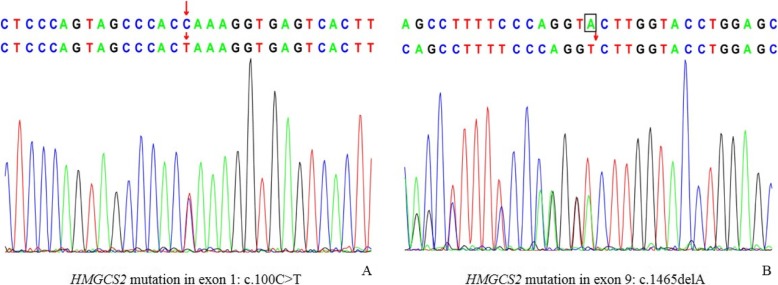
We report a 9-month-old boy with mHS deficiency presenting with unusually severe and persistent acidosis after diarrhea and reduced oral food intake. The metabolic acidosis persisted even after supplementation with sugar and alkaline solution. Blood purification and assisted respiration alleviated symptoms, but a second onset induced by respiratory infection several days later led to multiple organ failure and death. Urine organic acid analysis during the acute episode revealed a complex pattern of ketogenic dicarboxylic and 3-hydroxydicarboxylic aciduria with prominent elevation of glutaric acid and adipic acid, which seem to be specific to mHS deficiency. Plasma acylcarnitine analysis revealed elevated 3-hydroxybutyrylcarnitine and acetylcarnitine. This is the first report of elevated 3-hydroxybutyrylcarnitine in mHS deficiency. Whole exome sequencing revealed a novel compound heterozygous mutation in HMGCS2 (c.100C > T and c.1465delA).
This severe case suggests the need for patients with mHS deficiency to avoid recurrent illness because it can induce severe metabolic crisis, possibly leading to death. Such patients may also require special treatment, such as blood purification. Urine organic acid profile during the acute episode may give a hint to the disease.
线粒体 3-羟-3-甲基戊二酰辅酶 A 合酶(mHS)缺乏症是一种常染色体隐性遗传的代谢性疾病,会导致肝脏在患病或禁食期间酮体生成失败。这是一种非常罕见的疾病,全世界仅有少数病例报告,大多数患者在接受适当治疗后预后良好。
我们报告了一例 mHS 缺乏症患儿,在腹泻和减少口服食物摄入后出现异常严重和持续的酸中毒。代谢性酸中毒在补充糖和碱性溶液后仍持续存在。血液净化和辅助呼吸缓解了症状,但几天后因呼吸道感染再次发作导致多器官衰竭和死亡。急性发作期间的尿液有机酸分析显示出酮基二羧酸和 3-羟二羧酸尿症的复杂模式,伴有明显升高的戊二酸和己二酸,这似乎是 mHS 缺乏症的特异性表现。血浆酰基肉碱分析显示 3-羟基丁酰肉碱和乙酰肉碱升高。这是 mHS 缺乏症中首次报道 3-羟基丁酰肉碱升高。全外显子组测序显示 HMGCS2 中存在一种新型复合杂合突变(c.100C>T 和 c.1465delA)。
本例重症病例提示 mHS 缺乏症患者需要避免反复发作,因为这可能会引发严重的代谢危机,甚至导致死亡。此类患者可能还需要特殊治疗,如血液净化。急性发作期间的尿液有机酸谱可能提示该疾病。