School of Electrical Engineering and Computer Science, Washington State University, P.O. Box 642752, Pullman, Washington, USA.
Paul G. Allen School for Global Animal Health, Washington State University, P.O. Box 647090, Pullman, Washington, USA.
Sci Rep. 2019 Oct 9;9(1):14487. doi: 10.1038/s41598-019-50686-z.
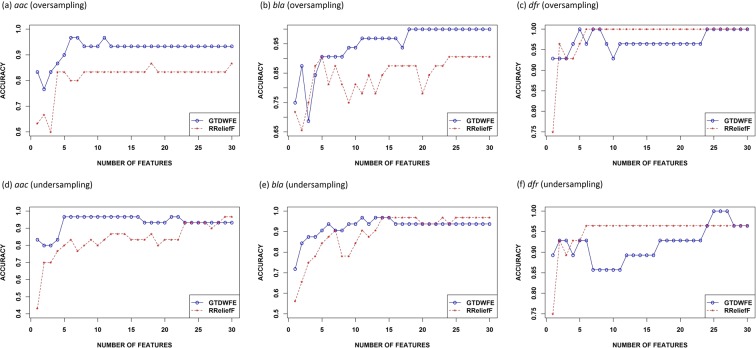
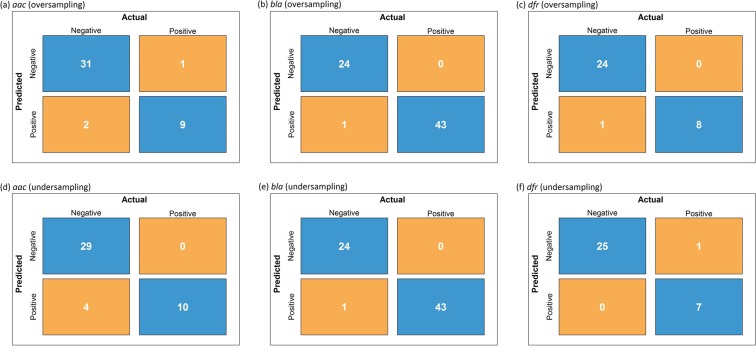
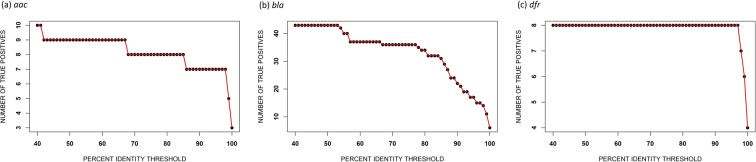
The increasing prevalence of antimicrobial-resistant bacteria drives the need for advanced methods to identify antimicrobial-resistance (AMR) genes in bacterial pathogens. With the availability of whole genome sequences, best-hit methods can be used to identify AMR genes by differentiating unknown sequences with known AMR sequences in existing online repositories. Nevertheless, these methods may not perform well when identifying resistance genes with sequences having low sequence identity with known sequences. We present a machine learning approach that uses protein sequences, with sequence identity ranging between 10% and 90%, as an alternative to conventional DNA sequence alignment-based approaches to identify putative AMR genes in Gram-negative bacteria. By using game theory to choose which protein characteristics to use in our machine learning model, we can predict AMR protein sequences for Gram-negative bacteria with an accuracy ranging from 93% to 99%. In order to obtain similar classification results, identity thresholds as low as 53% were required when using BLASTp.
抗菌药物耐药性细菌的不断增加,促使我们需要先进的方法来鉴定细菌病原体中的抗菌药物耐药性(AMR)基因。随着全基因组序列的出现,可以使用最佳比对方法通过区分未知序列与现有在线存储库中已知 AMR 序列来鉴定 AMR 基因。然而,当鉴定与已知序列具有低序列同一性的耐药基因时,这些方法的性能可能不佳。我们提出了一种机器学习方法,该方法使用蛋白质序列(序列同一性在 10% 到 90% 之间)作为替代传统 DNA 序列比对方法的方法,用于鉴定革兰氏阴性菌中的推定 AMR 基因。通过使用博弈论来选择在我们的机器学习模型中使用哪些蛋白质特征,我们可以预测革兰氏阴性菌的 AMR 蛋白质序列,准确率在 93% 到 99% 之间。为了获得类似的分类结果,当使用 BLASTp 时,需要将同一性阈值降低到低至 53%。