Marsico Lung Institute/Cystic Fibrosis Research Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina.
Division of Pediatric Pulmonology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina.
Pediatr Pulmonol. 2019 Nov;54 Suppl 3(Suppl 3):S84-S96. doi: 10.1002/ppul.24530.
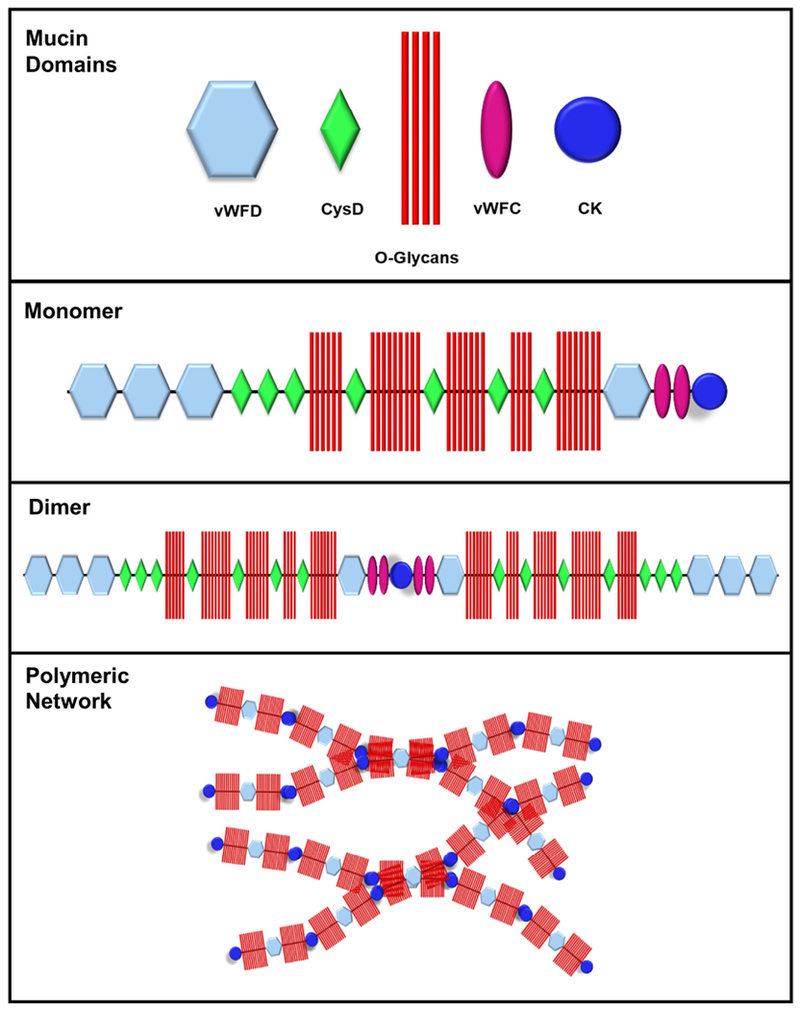
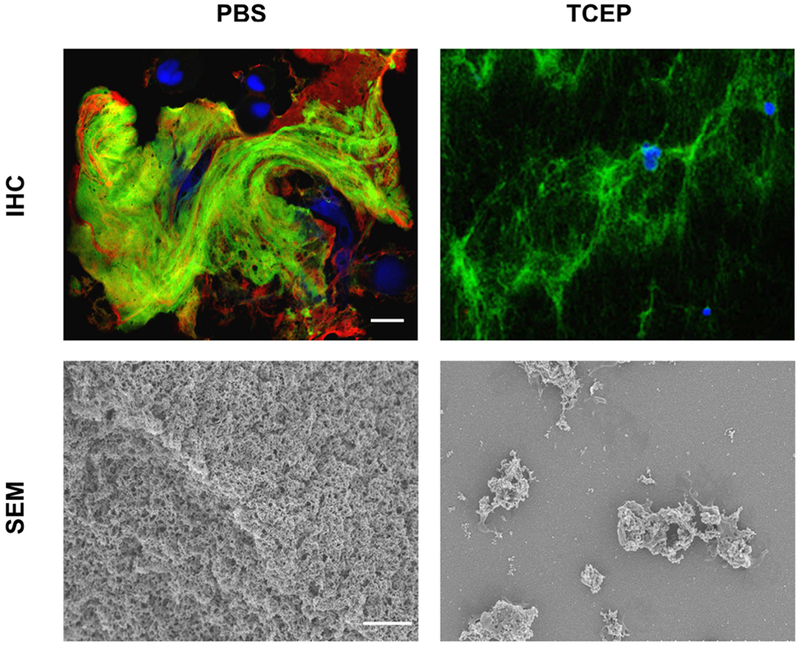
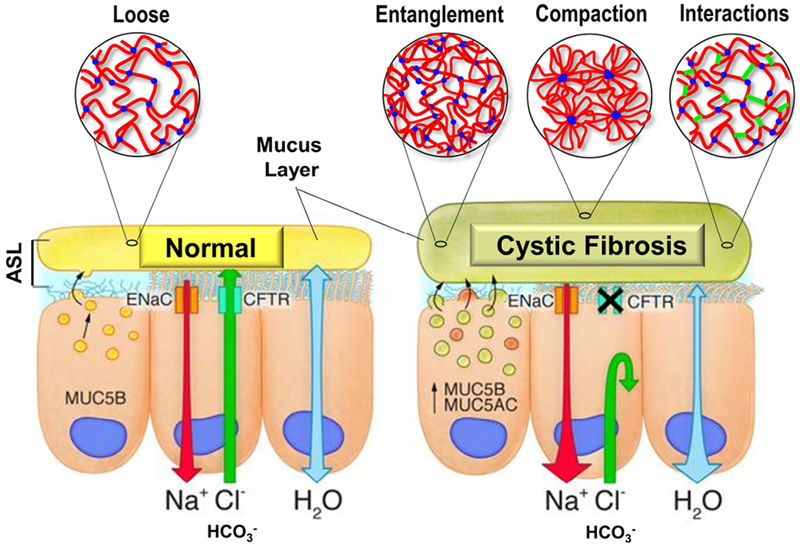
Cystic fibrosis (CF) is both the most common and most lethal genetic disease in the Caucasian population. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and is characterized by the accumulation of thick, adherent mucus plaques in multiple organs, of which the lungs, gastrointestinal tract and pancreatic ducts are the most commonly affected. A similar pathogenesis cascade is observed in all of these organs: loss of CFTR function leads to altered ion transport, consisting of decreased chloride and bicarbonate secretion via the CFTR channel and increased sodium absorption via epithelial sodium channel upregulation. Mucosa exposed to changes in ionic concentrations sustain severe pathophysiological consequences. Altered mucus biophysical properties and weakened innate defense mechanisms ensue, furthering the progression of the disease. Mucins, the high-molecular-weight glycoproteins responsible for the viscoelastic properties of the mucus, play a key role in the disease but the actual mechanism of mucus accumulation is still undetermined. Multiple hypotheses regarding the impact of CFTR malfunction on mucus have been proposed and are reviewed here. (a) Dehydration increases mucin monomer entanglement, (b) defective Ca chelation compromises mucin expansion, (c) ionic changes alter mucin interactions, and (d) reactive oxygen species increase mucin crosslinking. Although one biochemical change may dominate, it is likely that all of these mechanisms play some role in the progression of CF disease. This article discusses recent findings on the initial cause(s) of aberrant mucus properties in CF and examines therapeutic approaches aimed at correcting mucus properties.
囊性纤维化(CF)是白种人群体中最常见和最致命的遗传疾病。CF 是由囊性纤维化跨膜电导调节因子(CFTR)基因突变引起的,其特征是在多个器官中积聚厚厚的、黏附的黏液斑块,其中肺部、胃肠道和胰腺管是最常受影响的器官。在所有这些器官中都观察到类似的发病机制级联:CFTR 功能丧失导致离子转运改变,包括 CFTR 通道中氯离子和碳酸氢盐分泌减少以及上皮钠通道上调导致钠离子吸收增加。暴露于离子浓度变化的黏膜会承受严重的病理生理后果。改变的黏液生物物理特性和减弱的先天防御机制随之而来,进一步促进了疾病的发展。黏蛋白是负责黏液粘弹性特性的高分子量糖蛋白,在疾病中起着关键作用,但黏液积聚的实际机制仍未确定。这里回顾了关于 CFTR 功能障碍对黏液影响的多种假说。(a) 脱水增加黏蛋白单体缠结,(b) 钙螯合缺陷损害黏蛋白扩张,(c) 离子变化改变黏蛋白相互作用,以及 (d) 活性氧增加黏蛋白交联。尽管一种生化变化可能占主导地位,但所有这些机制都可能在 CF 疾病的进展中发挥一定作用。本文讨论了 CF 中异常黏液特性的初始原因的最新发现,并研究了旨在纠正黏液特性的治疗方法。