Department of Bacterial Infections, Research Institute for Microbial Diseases, Osaka University, Osaka, 565-0871, Japan.
Department of Infection Metagenomics, Research Institute for Microbial Diseases, Osaka University, Osaka, 565-0871, Japan.
Funct Integr Genomics. 2020 Jul;20(4):523-536. doi: 10.1007/s10142-020-00732-1. Epub 2020 Jan 18.
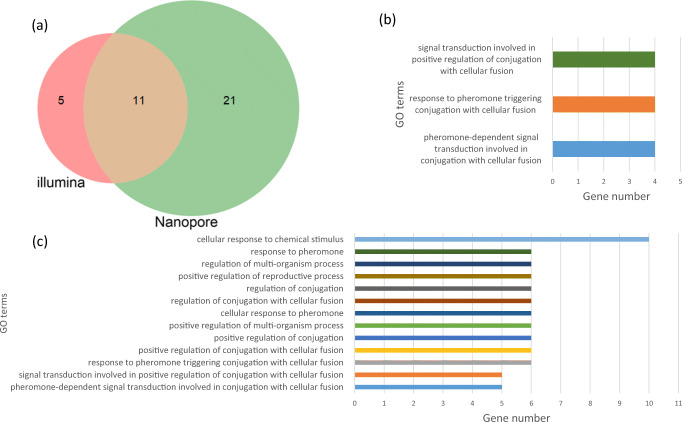
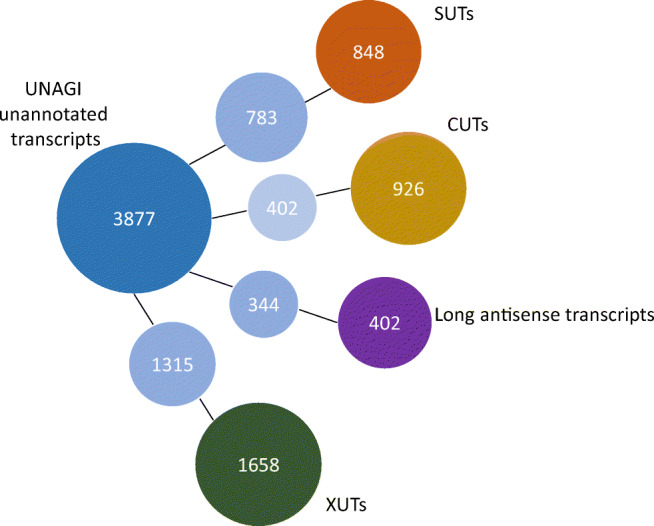
Sequencing the entire RNA molecule leads to a better understanding of the transcriptome architecture. SMARTer (Switching Mechanism at 5'-End of RNA Template) is a technology aimed at generating full-length cDNA from low amounts of mRNA for sequencing by short-read sequencers such as those from Illumina. However, short read sequencing such as Illumina technology includes fragmentation that results in bias and information loss. Here, we built a pipeline, UNAGI or UNAnnotated Gene Identifier, to process long reads obtained with nanopore sequencing and compared this pipeline with the standard Illumina pipeline by studying the Saccharomyces cerevisiae transcriptome in full-length cDNA samples generated from two different biological samples: haploid and diploid cells. Additionally, we processed the long reads with another long read tool, FLAIR. Our strand-aware method revealed significant differential gene expression that was masked in Illumina data by antisense transcripts. Our pipeline, UNAGI, outperformed the Illumina pipeline and FLAIR in transcript reconstruction (sensitivity and specificity of 80% and 40% vs. 18% and 34% and 79% and 32%, respectively). Moreover, UNAGI discovered 3877 unannotated transcripts including 1282 intergenic transcripts while the Illumina pipeline discovered only 238 unannotated transcripts. For isoforms profiling, UNAGI also outperformed the Illumina pipeline and FLAIR in terms of sensitivity (91% vs. 82% and 63%, respectively). But the low accuracy of nanopore sequencing led to a closer gap in terms of specificity with Illumina pipeline (70% vs. 63%) and to a huge gap with FLAIR (70% vs 0.02%).
对整个 RNA 分子进行测序可以更好地了解转录组结构。SMARTer(RNA 模板 5' 端切换机制)是一种旨在从低量 mRNA 生成全长 cDNA 的技术,用于短读测序仪(如 Illumina 测序仪)测序。然而,Illumina 等短读测序技术包括片段化,这会导致偏差和信息丢失。在这里,我们构建了一个管道,UNAGI 或 UNAnnotated Gene Identifier,用于处理使用纳米孔测序获得的长读段,并通过研究来自两种不同生物样本(单倍体和二倍体细胞)的全长 cDNA 样本中的酿酒酵母转录组,将该管道与标准 Illumina 管道进行比较。此外,我们还使用另一种长读段工具 FLAIR 处理了长读段。我们的链感知方法揭示了显著的差异基因表达,这些表达被反义转录本掩盖在 Illumina 数据中。我们的 UNAGI 管道在转录重建方面优于 Illumina 管道和 FLAIR(敏感性和特异性分别为 80%和 40%、18%和 34%、79%和 32%)。此外,UNAGI 发现了 3877 个未注释的转录本,包括 1282 个基因间转录本,而 Illumina 管道仅发现了 238 个未注释的转录本。对于异构体分析,UNAGI 在敏感性方面也优于 Illumina 管道和 FLAIR(91%、82%和 63%)。但是纳米孔测序的低准确性导致其特异性与 Illumina 管道的差距更小(70%对 63%),与 FLAIR 的差距更大(70%对 0.02%)。