Center for Molecular Medicine, Children's Hospital of Fudan University, Shanghai, China.
Key Laboratory of Birth Defects, Pediatrics Research Institute, Children's Hospital of Fudan University, Shanghai, China.
Mol Genet Genomic Med. 2020 Mar;8(3):e1144. doi: 10.1002/mgg3.1144. Epub 2020 Jan 27.
Congenital myasthenic syndrome 22 (CMS22) is a rare autosomal recessive disorder due to isolated PREPL deficiency and characterized by neonatal hypotonia, muscular weakness, and feeding difficulties. Eight such cases have already been reported, while maternal uniparental disomy with a PREPL pathogenic mutation has never been involved.
Trio whole-exome sequencing (WES), comparative genomic hybridization microarray (arry-CGH), and Sanger sequencing were performed on a 6-month-old girl with severe neonatal hypotonia and feeding difficulties. Also, the phenotype and genotype of reported CMS22 patients were reviewed.
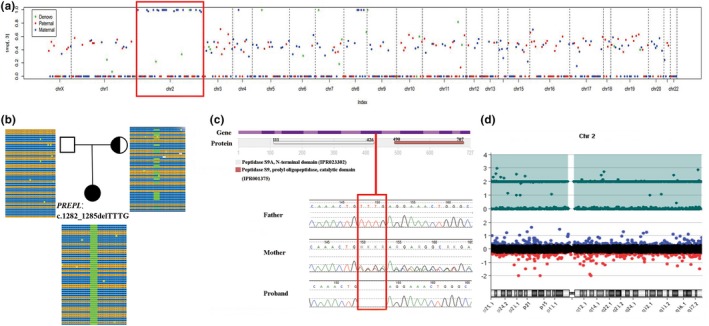
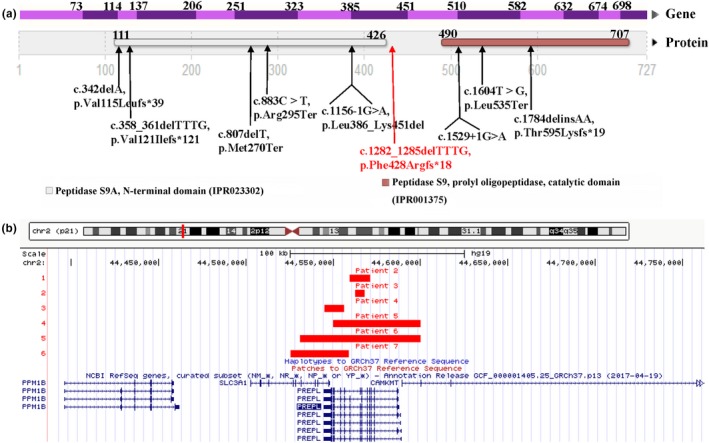
In this female infant, we identified a novel homozygous frameshift mutation in PREPL (c.1282_1285delTTTG, p.Phe428Argfs*18) by trio-WES. Sanger sequencing confirmed that her mother was heterozygous and her father was normal. Trio-WES data showed that 96.70% (1668/1725) variants on chromosome 2 were homozygous and maternally inherited, suggesting maternal uniparental disomy of chromosome 2 [UPD(2)mat]. Array-CGH did not show copy number variants (CNVs) but revealed complete UPD(2).
To date, nine patients with CMS22 have been reported including our patient, and we report the youngest and the first UPD(2)mat with PREPL novel homozygous pathogenic mutation case, which expand the mutation spectrum of PREPL gene.
先天性肌无力综合征 22 型(CMS22)是一种罕见的常染色体隐性遗传病,由 PREPL 基因的单一缺陷引起,其特征为新生儿肌无力、肌肉无力和喂养困难。目前已有 8 例此类病例报道,而 PREPL 致病突变的母体单亲二体性从未涉及。
对一名 6 月龄、严重新生儿肌无力和喂养困难的女孩进行了三体系列全外显子组测序(WES)、比较基因组杂交微阵列(array-CGH)和 Sanger 测序。同时,还回顾了已报道的 CMS22 患者的表型和基因型。
在这名女性婴儿中,我们通过三体系列 WES 发现了 PREPL 基因中的一个新的纯合移码突变(c.1282_1285delTTTG,p.Phe428Argfs*18)。Sanger 测序证实其母亲为杂合子,父亲正常。三体系列 WES 数据显示,2 号染色体上 96.70%(1668/1725)的变异为纯合且母源遗传,提示 2 号染色体的母体单亲二体性 [UPD(2)mat]。array-CGH 未显示拷贝数变异(CNVs),但显示完全 UPD(2)。
迄今为止,已有 9 例 CMS22 患者被报道,包括我们的患者,我们报告了首例携带 PREPL 基因新的纯合致病性突变的最年轻和首个 UPD(2)mat 病例,这扩展了 PREPL 基因突变谱。