The Jackson Laboratory for Genomic Medicine, 10 Discovery Drive, Farmington, CT, 06032, USA.
Oregon Health & Science University, Portland, OR, 97239, USA.
Orphanet J Rare Dis. 2020 Feb 4;15(1):40. doi: 10.1186/s13023-020-1313-0.
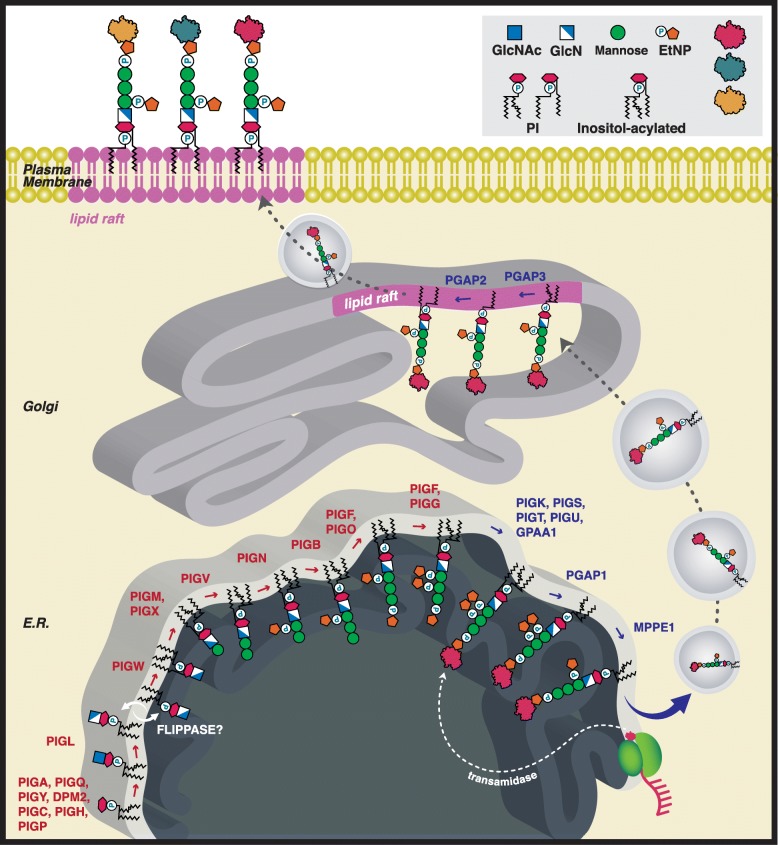
Defects in the glycosylphosphatidylinositol (GPI) biosynthesis pathway can result in a group of congenital disorders of glycosylation known as the inherited GPI deficiencies (IGDs). To date, defects in 22 of the 29 genes in the GPI biosynthesis pathway have been identified in IGDs. The early phase of the biosynthetic pathway assembles the GPI anchor (Synthesis stage) and the late phase transfers the GPI anchor to a nascent peptide in the endoplasmic reticulum (ER) (Transamidase stage), stabilizes the anchor in the ER membrane using fatty acid remodeling and then traffics the GPI-anchored protein to the cell surface (Remodeling stage).
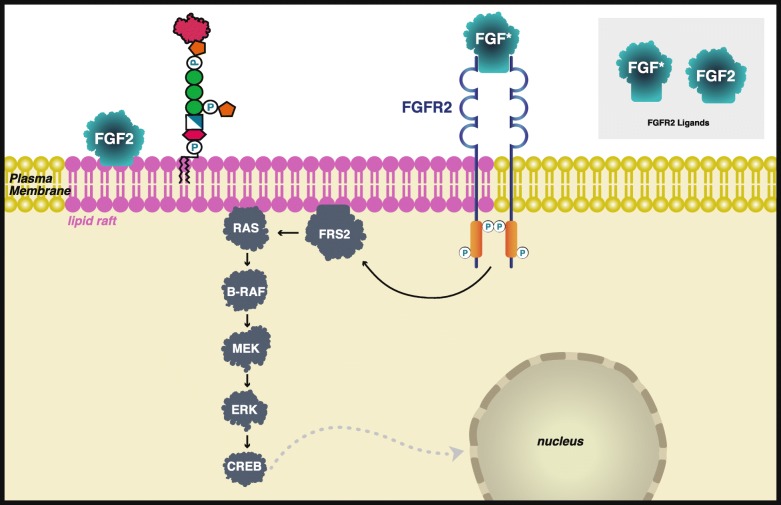
We addressed the hypothesis that disease-associated variants in either the Synthesis stage or Transamidase+Remodeling-stage GPI pathway genes have distinct phenotypic spectra. We reviewed clinical data from 58 publications describing 152 individual patients and encoded the phenotypic information using the Human Phenotype Ontology (HPO). We showed statistically significant differences between the Synthesis and Transamidase+Remodeling Groups in the frequencies of phenotypes in the musculoskeletal system, cleft palate, nose phenotypes, and cognitive disability. Finally, we hypothesized that phenotypic defects in the IGDs are likely to be at least partially related to defective GPI anchoring of their target proteins. Twenty-two of one hundred forty-two proteins that receive a GPI anchor are associated with one or more Mendelian diseases and 12 show some phenotypic overlap with the IGDs, represented by 34 HPO terms. Interestingly, GPC3 and GPC6, members of the glypican family of heparan sulfate proteoglycans bound to the plasma membrane through a covalent GPI linkage, are associated with 25 of these phenotypic abnormalities.
IGDs associated with Synthesis and Transamidase+Remodeling stages of the GPI biosynthesis pathway have significantly different phenotypic spectra. GPC2 and GPC6 genes may represent a GPI target of general disruption to the GPI biosynthesis pathway that contributes to the phenotypes of some IGDs.
糖基磷脂酰肌醇(GPI)生物合成途径的缺陷可导致一组称为遗传性 GPI 缺乏症(IGD)的先天性糖基化缺陷。迄今为止,在 IGD 中已鉴定出 GPI 生物合成途径的 29 个基因中的 22 个缺陷。生物合成途径的早期阶段组装 GPI 锚(合成阶段),晚期阶段将 GPI 锚转移到内质网(ER)中的新生肽(转酰胺酶阶段),使用脂肪酸重塑在 ER 膜中稳定锚,然后将 GPI 锚定蛋白转运到细胞表面(重塑阶段)。
我们提出了这样一个假设,即在合成阶段或转酰胺酶+重塑阶段 GPI 途径基因中,与疾病相关的变异具有不同的表型谱。我们回顾了 58 篇描述 152 名个体患者的临床数据,并使用人类表型本体论(HPO)对表型信息进行编码。我们表明,在骨骼肌肉系统、腭裂、鼻表型和认知障碍的表型频率方面,合成组和转酰胺酶+重塑组之间存在统计学上的显著差异。最后,我们假设 IGD 中的表型缺陷至少部分与靶蛋白的 GPI 锚定缺陷有关。在接受 GPI 锚定的 142 种蛋白质中,有 22 种与一种或多种孟德尔疾病相关,其中 12 种与 IGD 有一定的表型重叠,涉及 34 个 HPO 术语。有趣的是,GPC3 和 GPC6,硫酸乙酰肝素蛋白聚糖家族的成员,通过共价 GPI 连接与质膜结合,与其中 25 种表型异常有关。
与 GPI 生物合成途径的合成和转酰胺酶+重塑阶段相关的 IGD 具有显著不同的表型谱。GPC2 和 GPC6 基因可能代表 GPI 生物合成途径普遍破坏的 GPI 靶标,导致一些 IGD 的表型。