Department of Statistics, Oregon State University, Corvallis, OR 97330, USA.
Genes (Basel). 2020 Feb 10;11(2):185. doi: 10.3390/genes11020185.
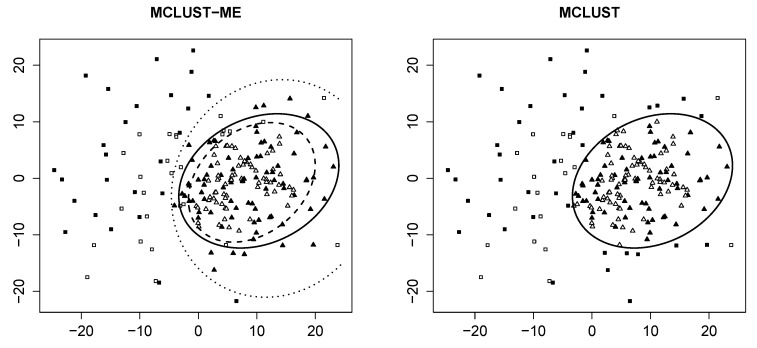
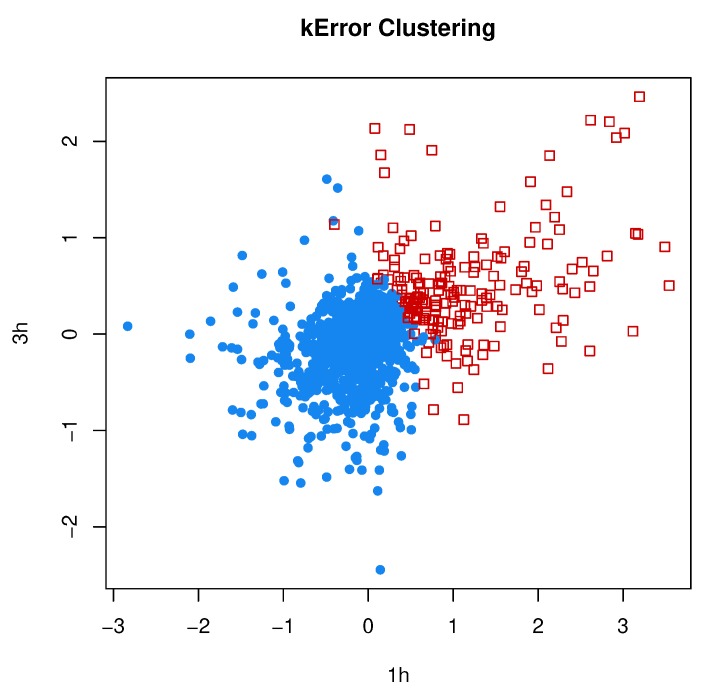
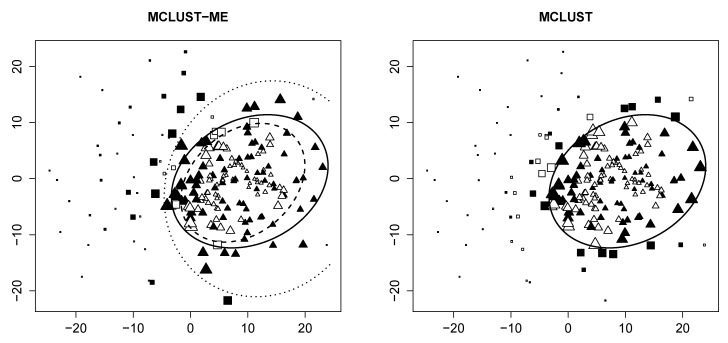
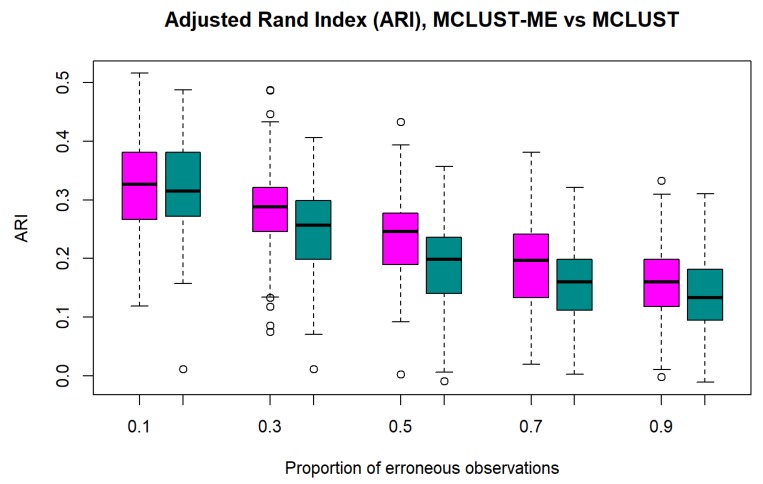
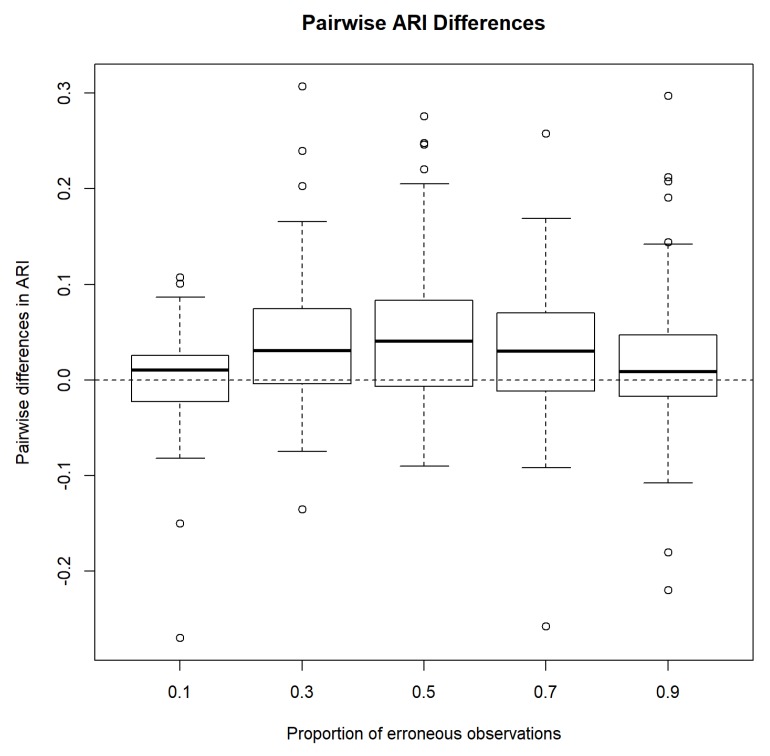
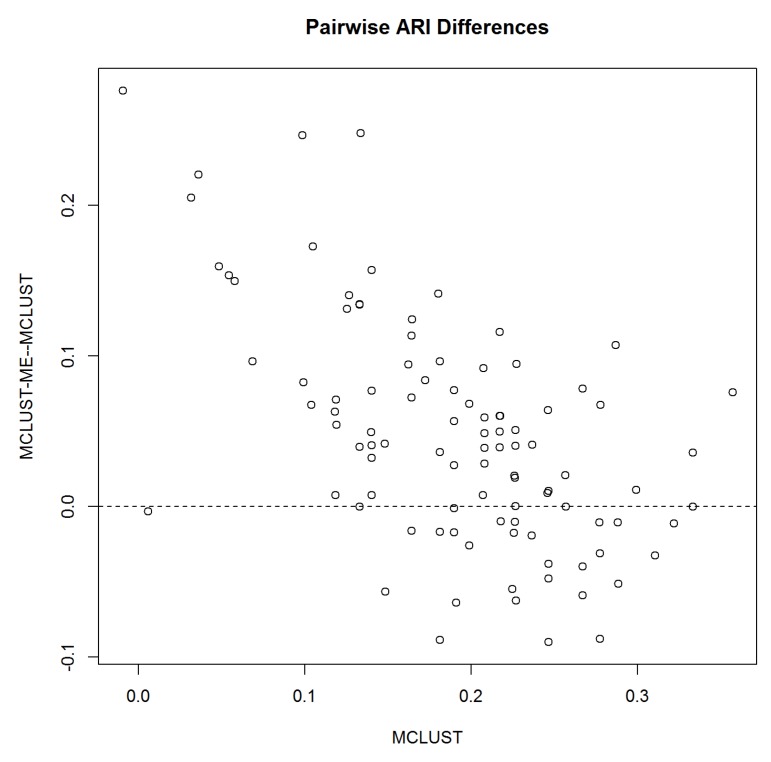
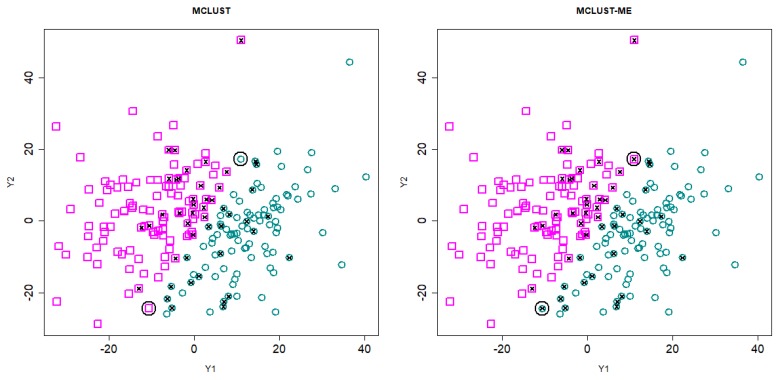
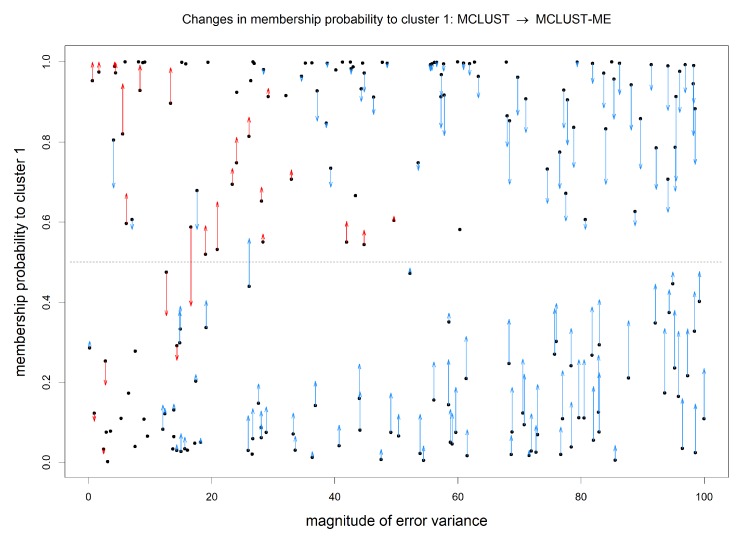
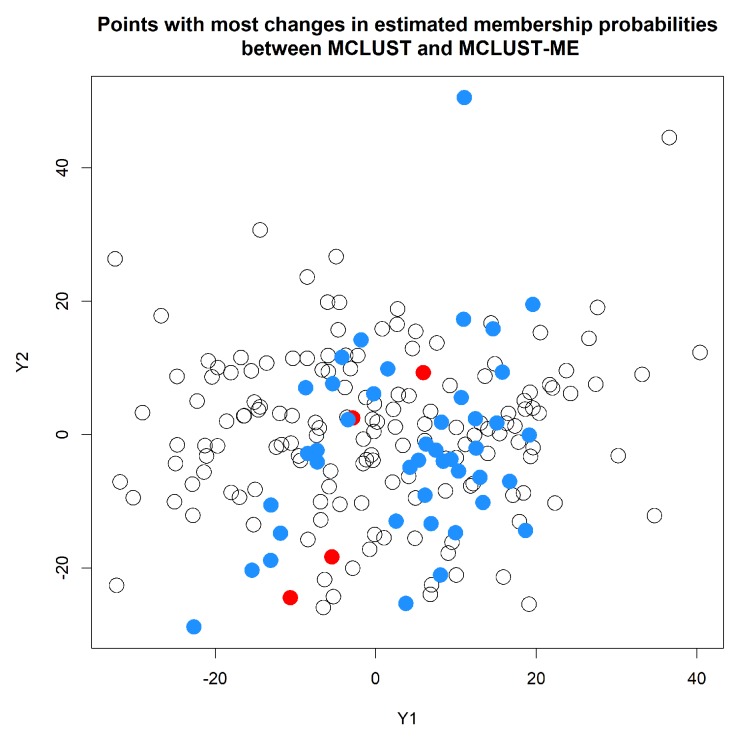
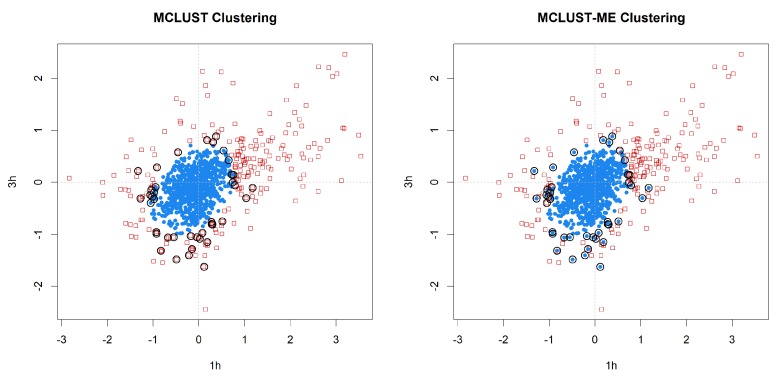
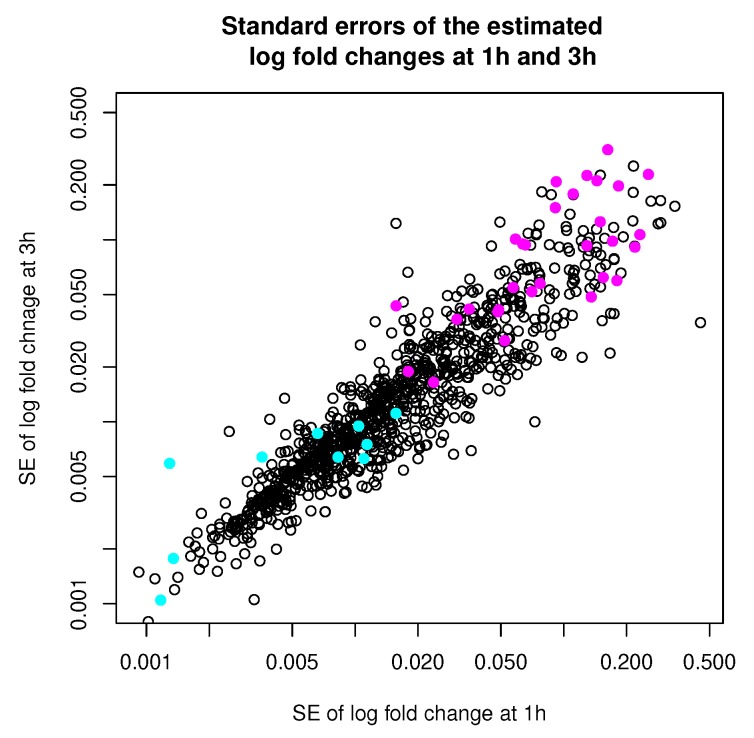
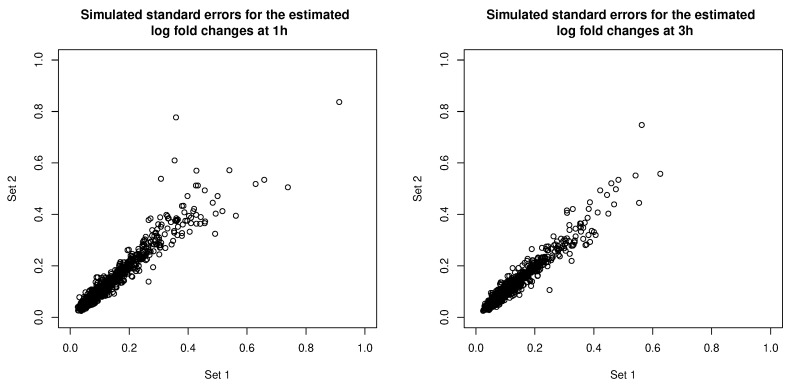
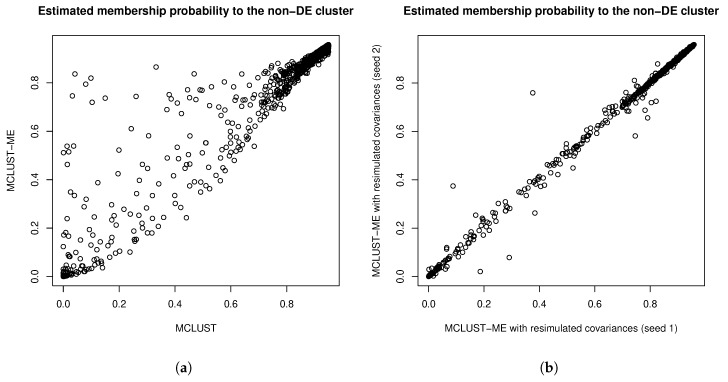
Model-based clustering with finite mixture models has become a widely used clustering method. One of the recent implementations is MCLUST. When objects to be clustered are summary statistics, such as regression coefficient estimates, they are naturally associated with estimation errors, whose covariance matrices can often be calculated exactly or approximated using asymptotic theory. This article proposes an extension to Gaussian finite mixture modeling-called MCLUST-ME-that properly accounts for the estimation errors. More specifically, we assume that the distribution of each observation consists of an underlying true component distribution and an independent measurement error distribution. Under this assumption, each unique value of estimation error covariance corresponds to its own classification boundary, which consequently results in a different grouping from MCLUST. Through simulation and application to an RNA-Seq data set, we discovered that under certain circumstances, explicitly, modeling estimation errors, improves clustering performance or provides new insights into the data, compared with when errors are simply ignored, whereas the degree of improvement depends on factors such as the distribution of error covariance matrices.
基于模型的聚类与有限混合模型已成为一种广泛使用的聚类方法。最近的实现之一是 MCLUST。当要聚类的对象是摘要统计信息(如回归系数估计)时,它们自然与估计误差相关联,其协方差矩阵通常可以使用渐近理论进行精确计算或近似计算。本文提出了对高斯有限混合模型的扩展,称为 MCLUST-ME,它可以正确地考虑到估计误差。更具体地说,我们假设每个观测值的分布由一个基本的真实分量分布和一个独立的测量误差分布组成。在这种假设下,每个独特的估计误差协方差值对应于其自己的分类边界,这就导致了与 MCLUST 不同的分组。通过模拟和对 RNA-Seq 数据集的应用,我们发现,在某些情况下,与简单忽略误差相比,明确地对估计误差进行建模可以提高聚类性能或为数据提供新的见解,而改进的程度取决于误差协方差矩阵的分布等因素。