Faculty of Physics, University of Vienna, Boltzmanngasse 5, 1090, Vienna, Austria.
Center for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo Miramon 182, 20014, San Sebastian, Spain.
Sci Rep. 2020 Feb 14;10(1):2684. doi: 10.1038/s41598-020-59401-9.
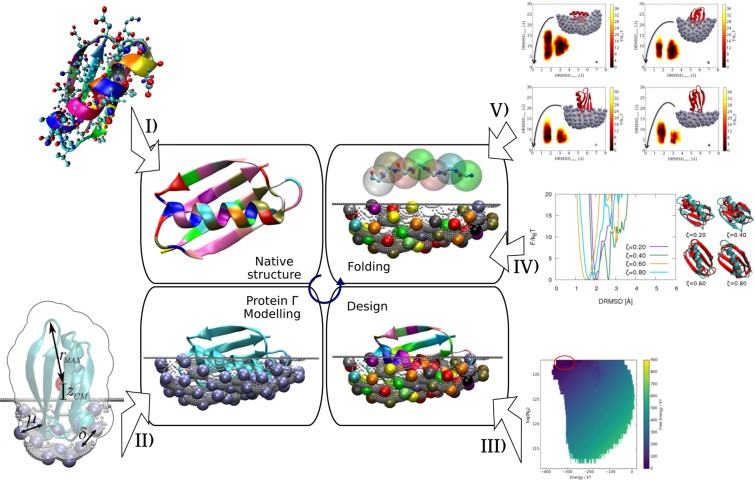
Isolating the properties of proteins that allow them to convert sequence into the structure is a long-lasting biophysical problem. In particular, studies focused extensively on the effect of a reduced alphabet size on the folding properties. However, the natural alphabet is a compromise between versatility and optimisation of the available resources. Here, for the first time, we include the impact of the relative availability of the amino acids to extract from the 20 letters the core necessary for protein stability. We present a computational protein design scheme that involves the competition for resources between a protein and a potential interaction partner that, additionally, gives us the chance to investigate the effect of the reduced alphabet on protein-protein interactions. We devise a scheme that automatically identifies the optimal reduced set of letters for the design of the protein, and we observe that even alphabets reduced down to 4 letters allow for single protein folding. However, it is only with 6 letters that we achieve optimal folding, thus recovering experimental observations. Additionally, we notice that the binding between the protein and a potential interaction partner could not be avoided with the investigated reduced alphabets. Therefore, we suggest that aggregation could have been a driving force in the evolution of the large protein alphabet.
将蛋白质的性质与其序列结构联系起来是一个长期存在的生物物理问题。特别是,研究广泛集中在减少字母表大小对折叠性质的影响。然而,自然字母表是在通用性和优化可用资源之间的妥协。在这里,我们首次考虑了从 20 个字母中提取蛋白质稳定性核心所需的氨基酸的相对可用性对其的影响。我们提出了一种计算蛋白质设计方案,该方案涉及蛋白质与潜在相互作用伙伴之间的资源竞争,这使我们有机会研究减少字母表对蛋白质-蛋白质相互作用的影响。我们设计了一种方案,可以自动确定设计蛋白质的最佳简化字母集,并且我们观察到,即使使用简化为 4 个字母的字母表,也可以实现单个蛋白质折叠。但是,只有使用 6 个字母才能实现最佳折叠,从而恢复了实验观察结果。此外,我们注意到,在所研究的简化字母表中,无法避免蛋白质与其潜在相互作用伙伴之间的结合。因此,我们建议,聚集可能是大蛋白质字母进化的驱动力。