Department of Psychiatry and Behavioral Neurosciences, University of Chicago, 924 East 57th Street Room. R016, Chicago, IL, 60637, USA.
Department of Psychiatry, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Mol Psychiatry. 2021 Jun;26(6):2048-2055. doi: 10.1038/s41380-020-0670-3. Epub 2020 Feb 17.
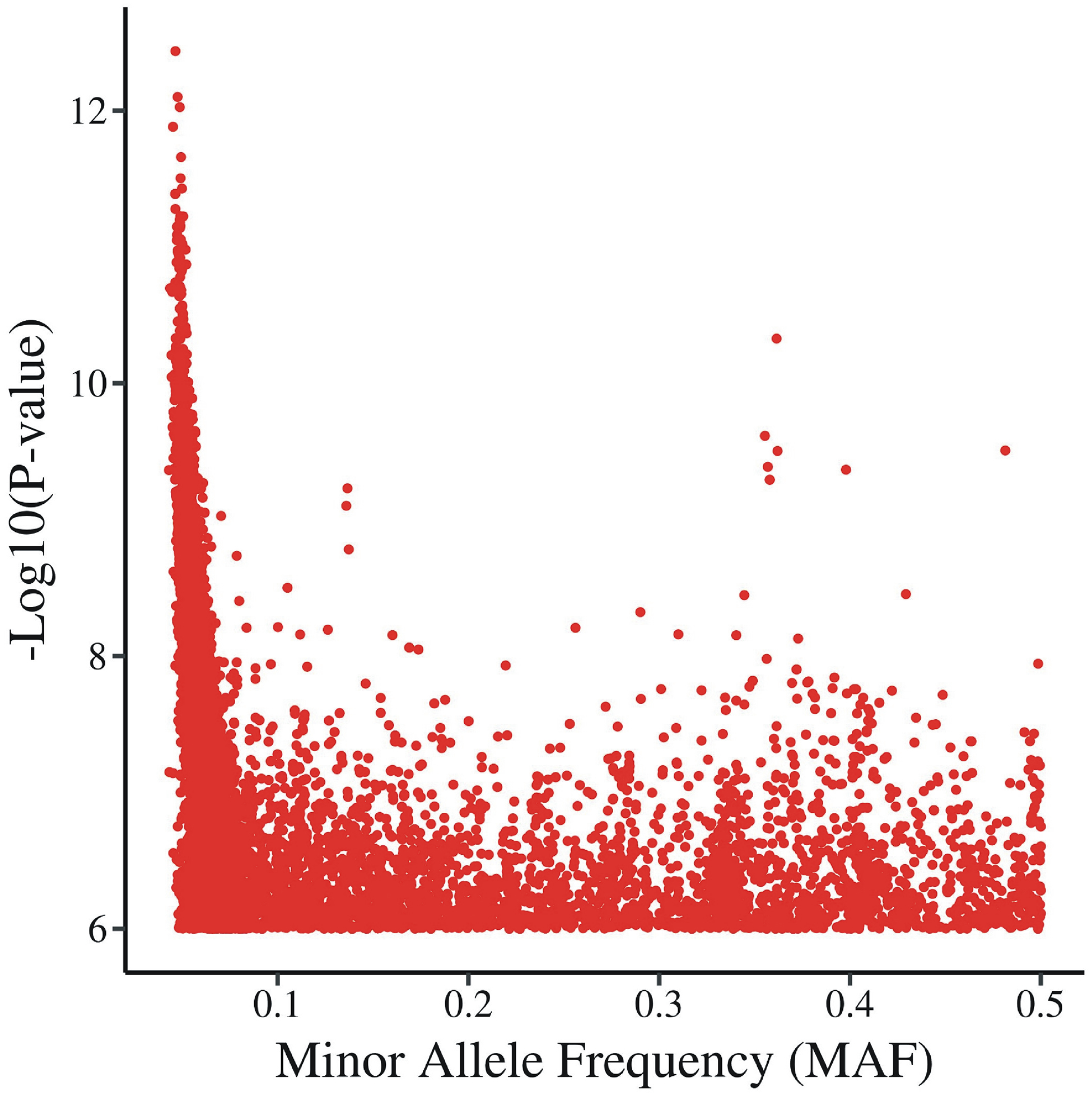
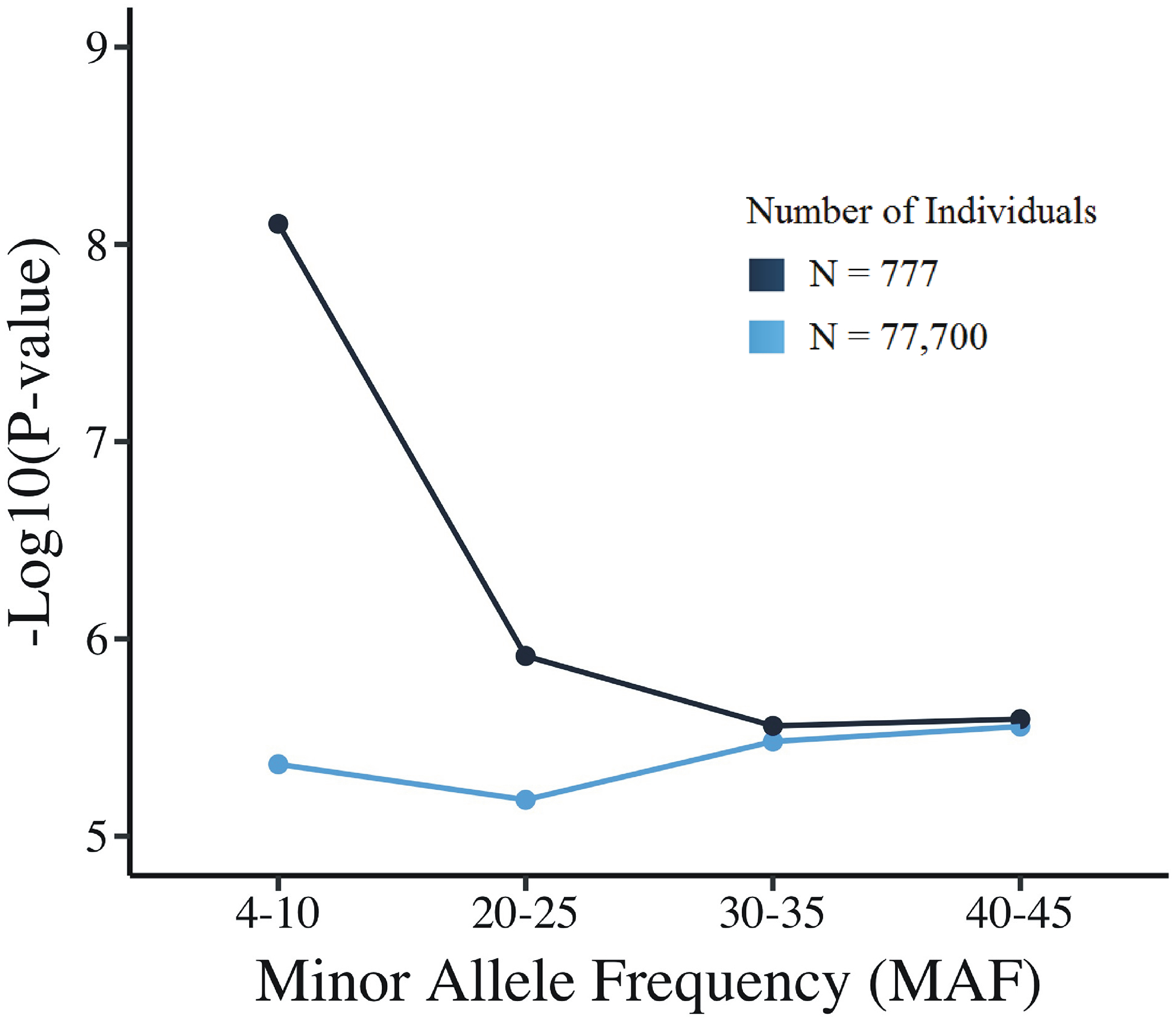
An important issue affecting genome-wide association studies with deep phenotyping (multiple correlated phenotypes) is determining the suitable family-wise significance threshold. Straightforward family-wise correction (Bonferroni) of p < 0.05 for 4.3 million genotypes and 335 phenotypes would give a threshold of p < 3.46E-11. This would be too conservative because it assumes all tests are independent. The effective number of tests, both phenotypic and genotypic, must be adjusted for the correlations between them. Spectral decomposition of the phenotype matrix and LD-based correction of the number of tested SNPs are currently used to determine an effective number of tests. In this paper, we compare these calculated estimates with permutation-determined family-wise significance thresholds. Permutations are performed by shuffling individual IDs of the genotype vector for this dataset, to preserve correlation of phenotypes. Our results demonstrate that the permutation threshold is influenced by minor allele frequency (MAF) of the SNPs, and by the number of individuals tested. For the more common SNPs (MAF > 0.1), the permutation family-wise threshold was in close agreement with spectral decomposition methods. However, for less common SNPs (0.05 < MAF ≤ 0.1), the permutation threshold calculated over all SNPs was off by orders of magnitude. This applies to the number of individuals studied (here 777) but not to very much larger numbers. Based on these findings, we propose that the threshold to find a particular level of family-wise significance may need to be established using separate permutations of the actual data for several MAF bins.
影响深度表型(多个相关表型)全基因组关联研究的一个重要问题是确定合适的总体显著水平阈值。对于 430 万个基因型和 335 个表型,直接进行简单的总体校正(Bonferroni),p < 0.05 的阈值将为 p < 3.46E-11。这将过于保守,因为它假设所有检验都是独立的。必须根据它们之间的相关性调整检验的有效数量,包括表型和基因型检验。目前,使用谱分解表型矩阵和基于 LD 的校正测试的 SNP 数量来确定有效检验数量。在本文中,我们将这些计算估计与置换确定的总体显著水平阈值进行了比较。置换是通过打乱该数据集基因型向量的个体 ID 来进行的,以保留表型的相关性。我们的结果表明,置换阈值受 SNP 的次要等位基因频率(MAF)和测试个体数量的影响。对于更常见的 SNP(MAF > 0.1),置换总体显著阈值与谱分解方法非常吻合。然而,对于不太常见的 SNP(0.05 < MAF ≤ 0.1),计算所有 SNP 的置换总体阈值相差几个数量级。这适用于研究的个体数量(此处为 777),但不适用于更大的数量。基于这些发现,我们提出,为了找到特定水平的总体显著,可能需要使用实际数据的不同置换来为几个 MAF 间隔建立阈值。