Laboratory of Diagnosis and Therapy of Lysosomal Disorders, Department of Women's and Children's Health, University of Padova, Via Giustiniani 3, 35128 Padova, Italy.
Fondazione Istituto di Ricerca Pediatrica "Città della Speranza", Corso Stati Uniti 4, 35127 Padova, Italy.
Int J Mol Sci. 2020 Feb 13;21(4):1258. doi: 10.3390/ijms21041258.
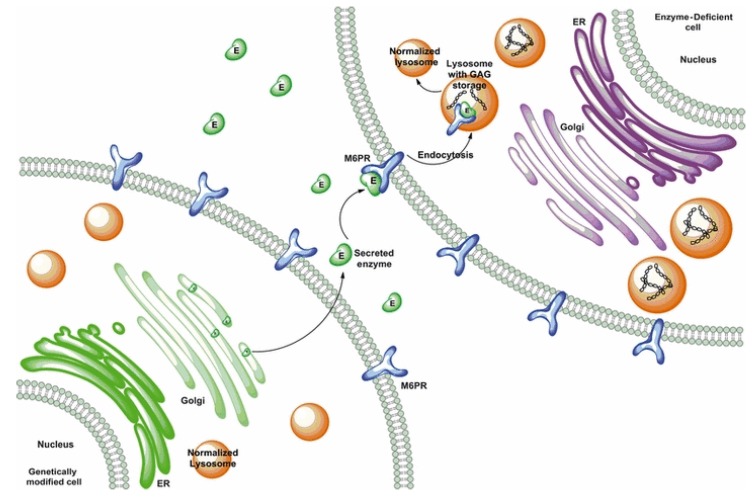
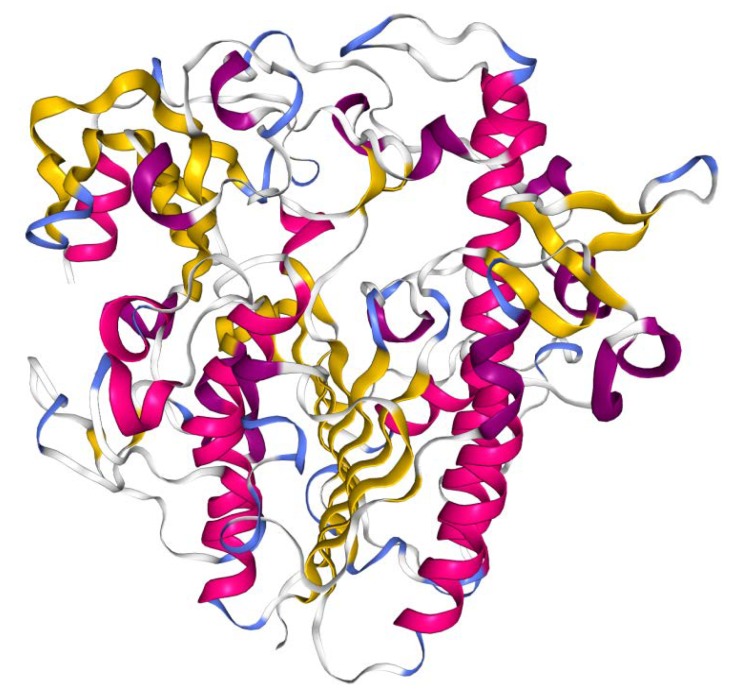
Mucopolysaccharidosis type II (MPS II, Hunter syndrome) was first described by Dr. Charles Hunter in 1917. Since then, about one hundred years have passed and Hunter syndrome, although at first neglected for a few decades and afterwards mistaken for a long time for the similar disorder Hurler syndrome, has been clearly distinguished as a specific disease since 1978, when the distinct genetic causes of the two disorders were finally identified. MPS II is a rare genetic disorder, recently described as presenting an incidence rate ranging from 0.38 to 1.09 per 100,000 live male births, and it is the only X-linked-inherited mucopolysaccharidosis. The complex disease is due to a deficit of the lysosomal hydrolase iduronate 2-sulphatase, which is a crucial enzyme in the stepwise degradation of heparan and dermatan sulphate. This contributes to a heavy clinical phenotype involving most organ-systems, including the brain, in at least two-thirds of cases. In this review, we will summarize the history of the disease during this century through clinical and laboratory evaluations that allowed its definition, its correct diagnosis, a partial comprehension of its pathogenesis, and the proposition of therapeutic protocols. We will also highlight the main open issues related to the possible inclusion of MPS II in newborn screenings, the comprehension of brain pathogenesis, and treatment of the neurological compartment.
黏多糖贮积症 II 型(MPS II,亨特综合征)于 1917 年由查尔斯·亨特博士首次描述。此后,大约一百年过去了,尽管亨特综合征起初被忽视了几十年,后来又被误诊为类似的 Hurler 综合征很长一段时间,但自 1978 年两种疾病的独特遗传原因最终被确定以来,它已被明确区分成一种特定的疾病。MPS II 是一种罕见的遗传疾病,最近的描述表明其发病率在每 10 万活产男婴中有 0.38 至 1.09 例,是唯一一种 X 连锁遗传的黏多糖贮积症。这种复杂的疾病是由于溶酶体水解酶艾杜糖 2-硫酸酯酶的缺乏引起的,该酶是肝素和硫酸皮肤素逐步降解过程中的关键酶。这导致了严重的临床表现,涉及到至少三分之二的病例中的大多数器官系统,包括大脑。在这篇综述中,我们将通过临床和实验室评估来总结这个世纪以来该病的历史,这些评估使得该病得以定义、正确诊断、对其发病机制有了部分理解,并提出了治疗方案。我们还将强调与可能将 MPS II 纳入新生儿筛查、理解脑发病机制和治疗神经学方面相关的主要问题。