Respiratory Department, Tianjin Medical University General Hospital, Tianjin, 300052, China.
Neurobiology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, 27709, USA.
J Neuroinflammation. 2020 Feb 18;17(1):64. doi: 10.1186/s12974-020-1728-5.
Sepsis-associated acute brain inflammation, if unresolved, may cause chronic neuroinflammation and resultant neurodegenerative diseases. However, little is known how the transition from acute to chronic neuroinflammation, which is critical for the following progressive neurodegeneration, occurs in sepsis. The goal of this study was to investigate potential immune factors regulating the transition process using a widely used endotoxemia LPS mouse model. This model shows distinct acute and chronic phases of neuroinflammation and recapitulates many cardinal features of Parkinson's disease, thus, providing a unique opportunity for studying phase transition of neuroinflammation.
C57BL/6 J, NLRP3, and IL-1R1 mice were employed. Mild and severe endotoxemia were produced by LPS ip injection at 1 or 5 mg/kg. Neuroinflammation in vitro and in vivo was assessed with proinflammatory cytokine expression by qPCR or ELISA and microglial activation by immunohistochemical analysis. Neurodegeneration was measured by manual and stereological counts of nigral dopaminergic neurons and immunohistochemical analysis of protein nitrosylation and α-synuclein phosphorylation.
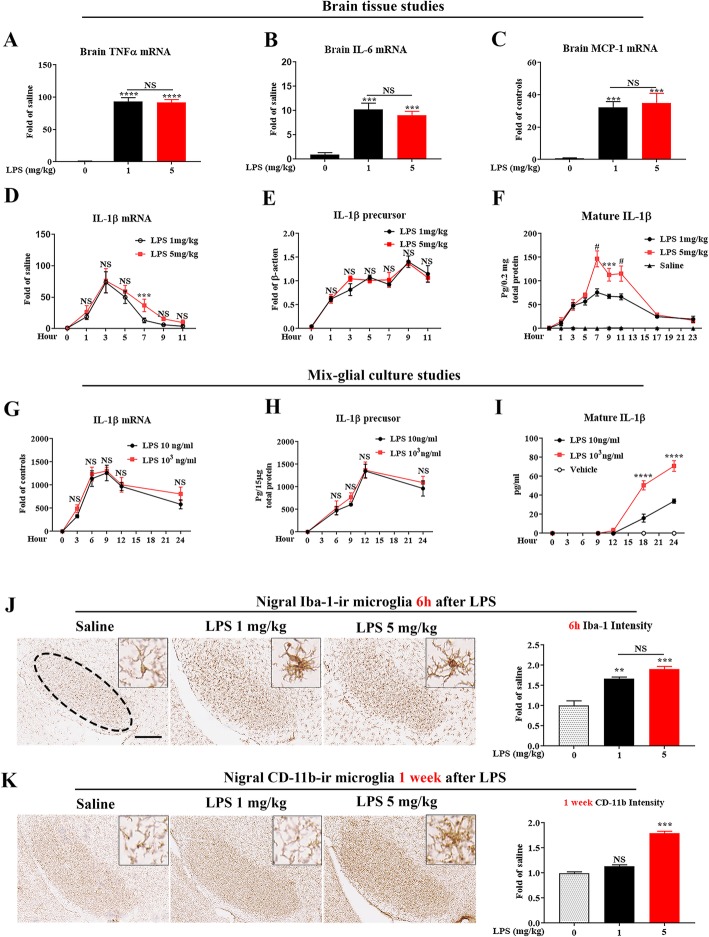
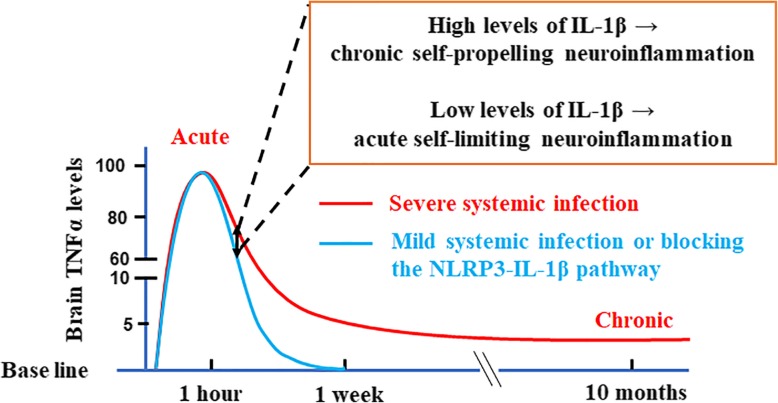
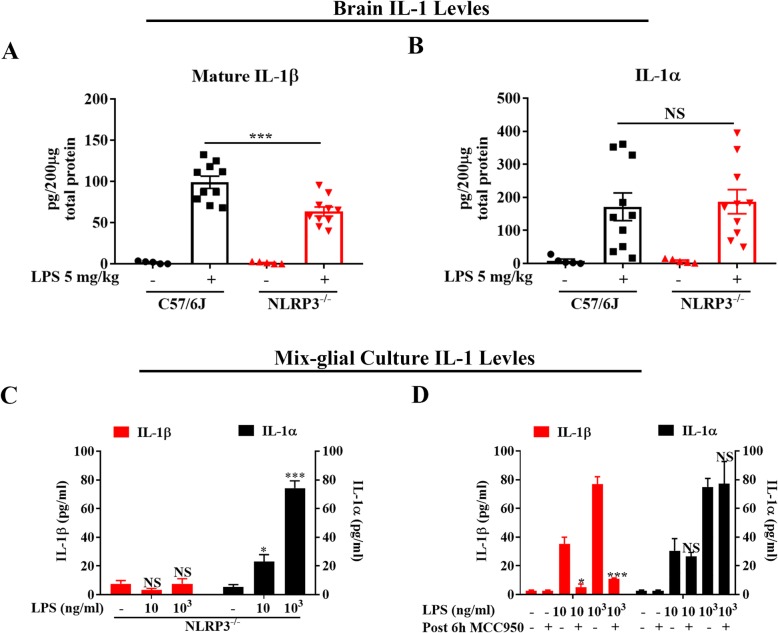
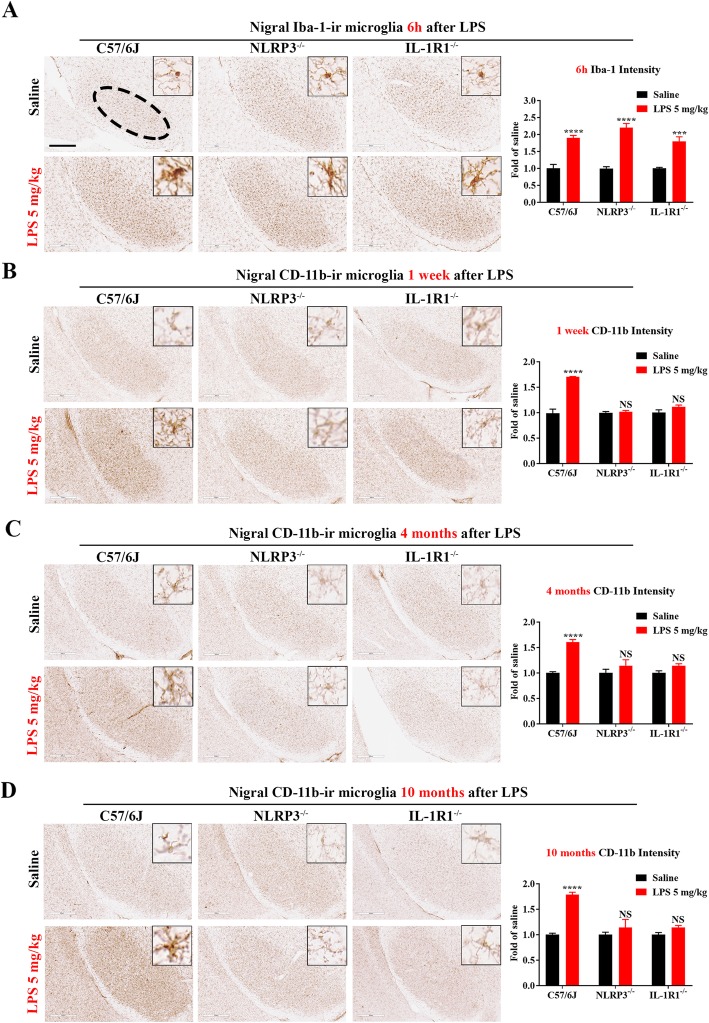
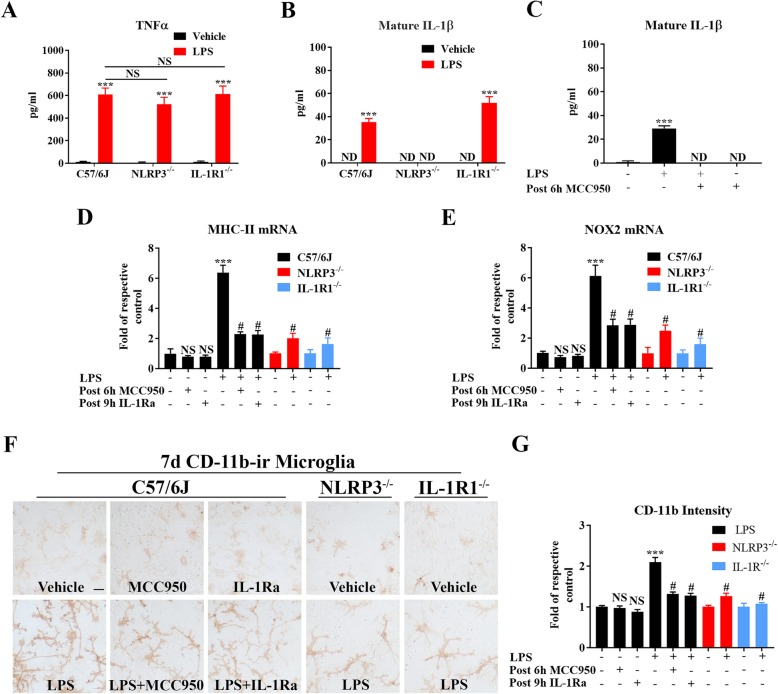
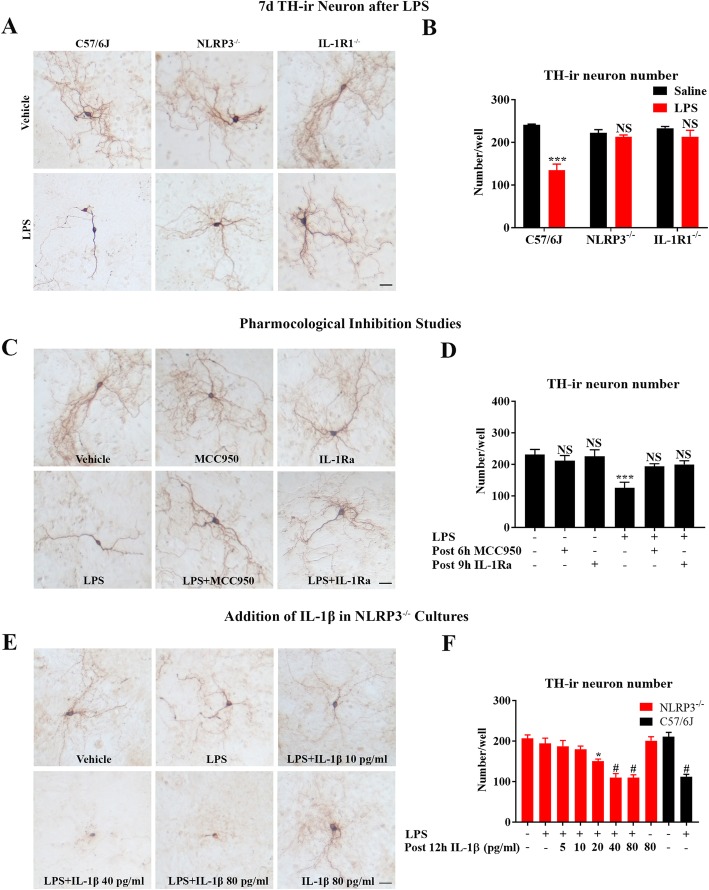
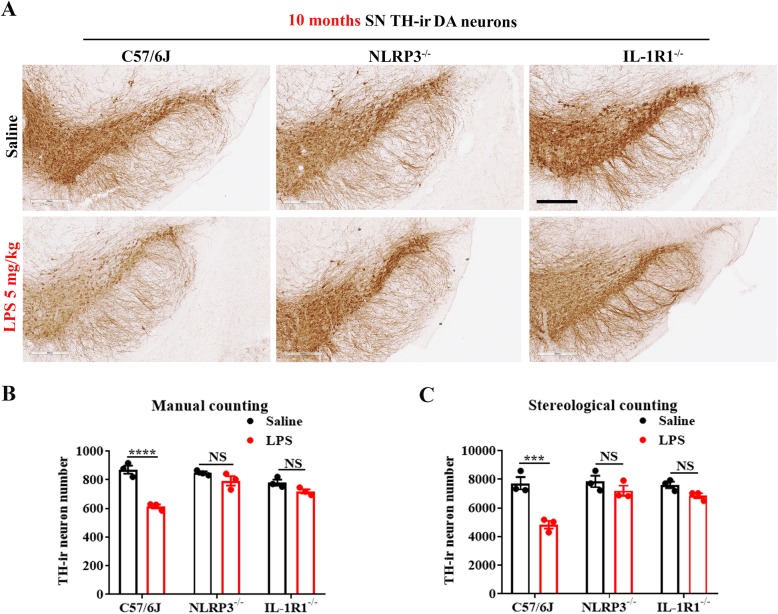
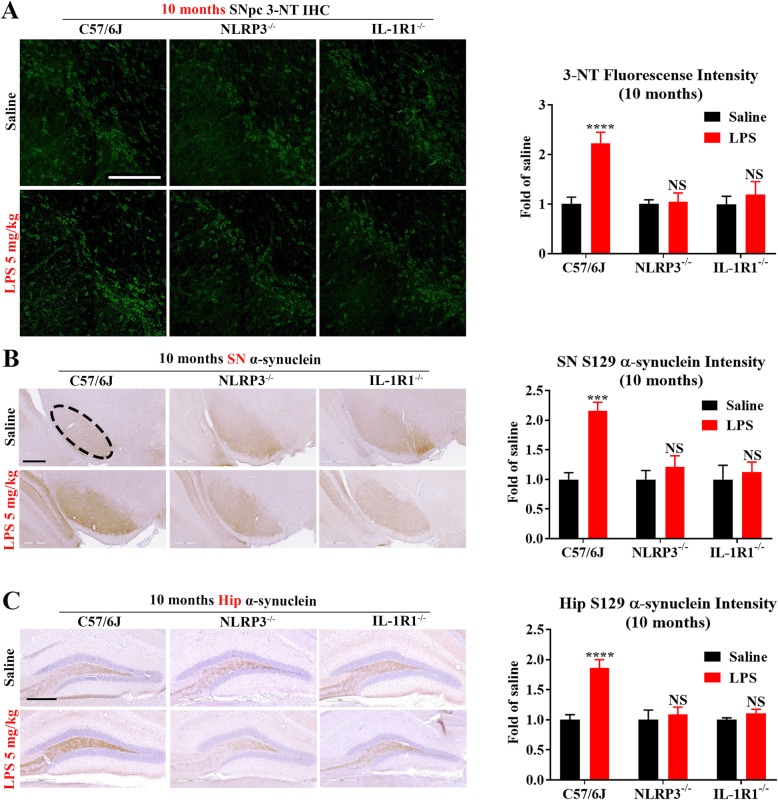
LPS-elicited initial increases in mouse brain mRNA levels of TNFα, IL-6, IL-1β, and MCP-1, and nigral microglial activation were not dose-related. By contrast, the delayed increase in brain mature IL-1β levels was dependent on LPS doses and protracted nigral microglial activation was only observed in high dose of LPS-treated mice. LPS-elicited increase in brain mature IL-1β but not IL-1α level was NLRP3-dependent. After high dose LPS treatment, deficiency of NLRP3 or IL-1R1 did not prevent the initiation of acute neuroinflammation but abolished chronic neuroinflammation. Genetic or pharmacological inhibition of the NLRP3-IL-1β axis repressed LPS-stimulated upregulation of chronic neuroinflammatory mediators including MHC-II, NOX2, and Mac1, and protected dopaminergic neurons. Ten months after LPS-elicited severe endotoxemia, nigral persisted microglial activation, elevated nitrosylated proteins and phosphorylated α-synuclein, and significant neuronal degeneration developed in wild-type mice but not in NLRP3 or IL-1R1 mice.
This study uncovers a novel role of the NLRP3-IL-1β signaling pathway in gauging the severity of sepsis-associated inflammation and determining whether acute neuroinflammation will resolve or transition to low grade chronic neuroinflammation. These findings also provide novel targets for developing therapy for severe systemic infection-related neurodegeneration.
如果不加以解决,脓毒症相关的急性脑炎症可能会导致慢性神经炎症,并导致退行性神经疾病。然而,人们对于从急性炎症向慢性炎症的转变过程知之甚少,而这对于随后的进行性神经退行性变至关重要。本研究旨在使用广泛应用的内毒素血症 LPS 小鼠模型,研究潜在的免疫因子如何调节这一转变过程。该模型显示出明显的急性和慢性神经炎症阶段,并再现了帕金森病的许多主要特征,因此为研究神经炎症的阶段转变提供了独特的机会。
使用 C57BL/6J、NLRP3 和 IL-1R1 小鼠。通过 LPS 腹腔注射 1 或 5mg/kg 产生轻度和重度内毒素血症。通过 qPCR 或 ELISA 检测促炎细胞因子表达和免疫组织化学分析检测小胶质细胞激活,评估体外和体内的神经炎症。通过手动和立体计数黑质多巴胺能神经元和免疫组织化学分析蛋白硝化和α-突触核蛋白磷酸化来测量神经退行性变。
LPS 诱导的小鼠大脑 TNFα、IL-6、IL-1β 和 MCP-1 的 mRNA 水平最初增加,以及黑质小胶质细胞激活与剂量无关。相比之下,大脑成熟的 IL-1β 水平的延迟增加取决于 LPS 剂量,只有在高剂量 LPS 处理的小鼠中才观察到持续的黑质小胶质细胞激活。LPS 诱导的大脑成熟 IL-1β 而不是 IL-1α 水平的增加依赖于 NLRP3。在高剂量 LPS 处理后,NLRP3 或 IL-1R1 的缺乏并不能防止急性神经炎症的发生,但消除了慢性神经炎症。NLRP3-IL-1β 轴的遗传或药理学抑制抑制了 LPS 刺激的慢性神经炎症介质的上调,包括 MHC-II、NOX2 和 Mac1,并保护多巴胺能神经元。在 LPS 引起的严重内毒素血症 10 个月后,黑质持续存在小胶质细胞激活、升高的硝化蛋白和磷酸化的α-突触核蛋白,并且在野生型小鼠中但不是在 NLRP3 或 IL-1R1 小鼠中发展出显著的神经元变性。
本研究揭示了 NLRP3-IL-1β 信号通路在评估脓毒症相关炎症严重程度以及确定急性神经炎症是否会消退或转变为低度慢性神经炎症方面的新作用。这些发现还为开发治疗严重全身感染相关神经退行性变的新疗法提供了新的靶点。