Lu Hengyu, Liu Chen, Huynh Hung, Le Thi Bich Uyen, LaMarche Matthew J, Mohseni Morvarid, Engelman Jeffrey A, Hammerman Peter S, Caponigro Giordano, Hao Huai-Xiang
Novartis Institutes for Biomedical Research, Oncology Disease Area, Cambridge, Massachusetts, USA.
Laboratory of Molecular Endocrinology, Division of Molecular and Cellular Research, National Cancer Centre, Singapore.
Oncotarget. 2020 Jan 21;11(3):265-281. doi: 10.18632/oncotarget.27435.
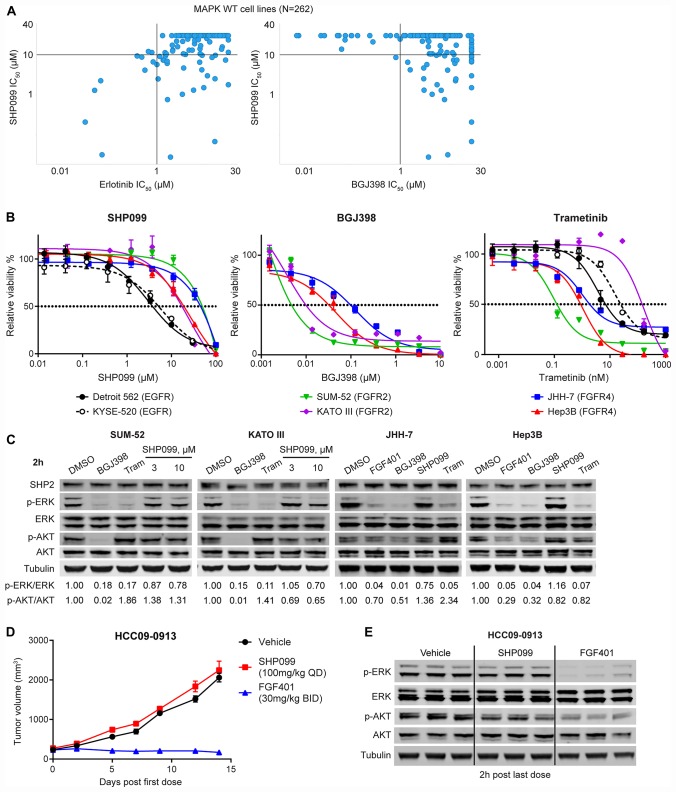
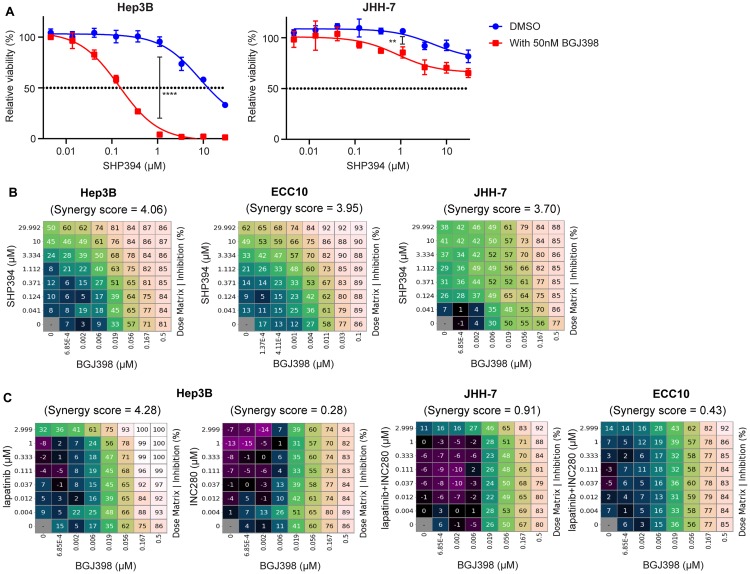
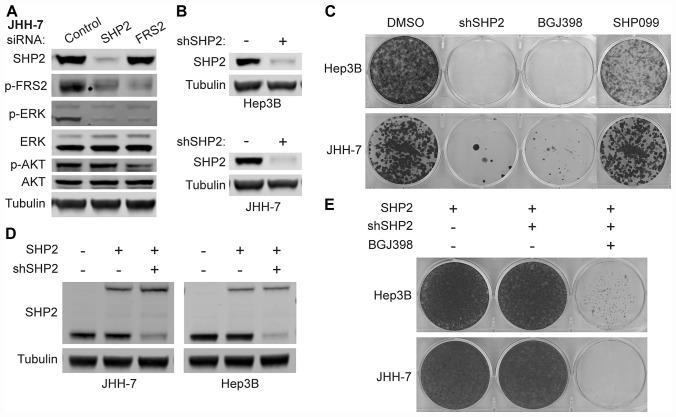
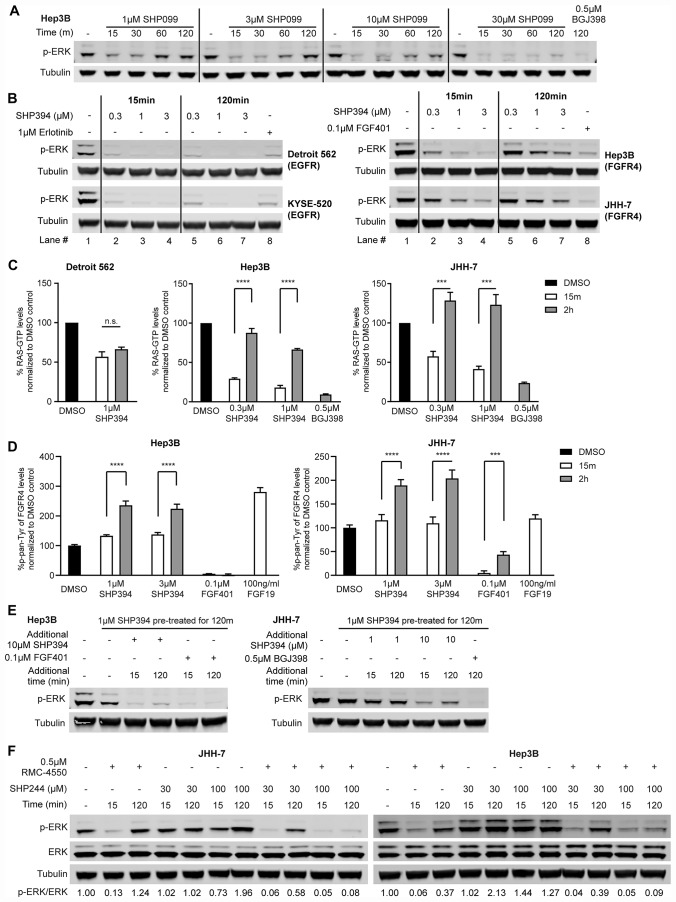
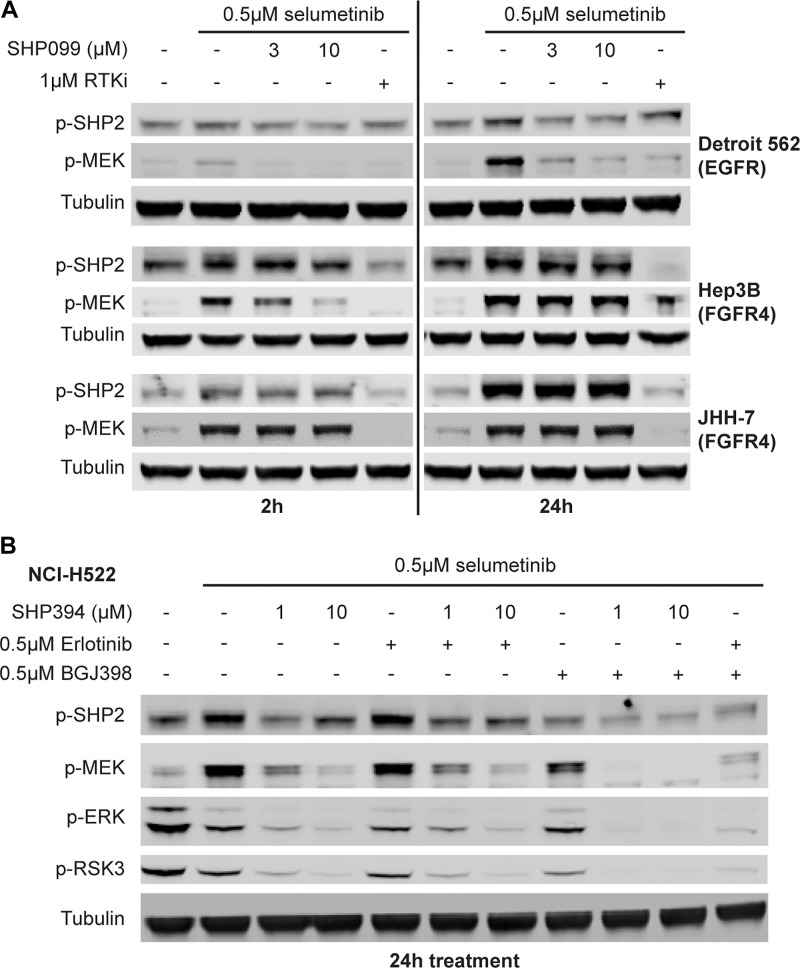
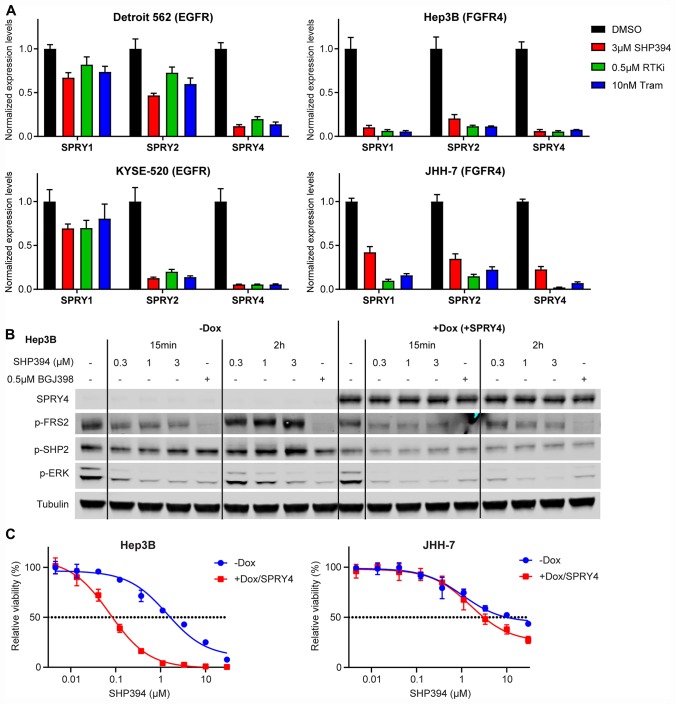
SHP2 mediates RAS activation downstream of multiple receptor tyrosine kinases (RTKs) and cancer cell lines dependent on RTKs are in general dependent on SHP2. Profiling of the allosteric SHP2 inhibitor SHP099 across cancer cell lines harboring various RTK dependencies reveals that FGFR-dependent cells are often insensitive to SHP099 when compared to EGFR-dependent cells. We find that FGFR-driven cells depend on SHP2 but exhibit resistance to SHP2 inhibitors and . Treatment of such models with SHP2 inhibitors results in an initial decrease in phosphorylated ERK1/2 (p-ERK) levels, however p-ERK levels rapidly rebound within two hours. This p-ERK rebound is blocked by FGFR inhibitors or high doses of SHP2 inhibitors. Mechanistically, compared with EGFR-driven cells, FGFR-driven cells tend to express high levels of RTK negative regulators such as the SPRY family proteins, which are rapidly downregulated upon ERK inhibition. Moreover, over-expression of SPRY4 in FGFR-driven cells prevents MAPK pathway reactivation and sensitizes them to SHP2 inhibitors. We also identified two novel combination approaches to enhance the efficacy of SHP2 inhibitors, either with a distinct site 2 allosteric SHP2 inhibitor or with a RAS-SOS1 interaction inhibitor. Our findings suggest the rapid FGFR feedback activation following initial pathway inhibition by SHP2 inhibitors may promote the open conformation of SHP2 and lead to resistance to SHP2 inhibitors. These findings may assist to refine patient selection and predict resistance mechanisms in the clinical development of SHP2 inhibitors and to suggest strategies for discovering SHP2 inhibitors that are more effective against upstream feedback activation.
SHP2介导多种受体酪氨酸激酶(RTK)下游的RAS激活,依赖RTK的癌细胞系通常也依赖SHP2。对携带各种RTK依赖性的癌细胞系进行变构SHP2抑制剂SHP099的分析发现,与依赖EGFR的细胞相比,依赖FGFR的细胞通常对SHP099不敏感。我们发现,由FGFR驱动的细胞依赖SHP2,但对SHP2抑制剂表现出抗性。用SHP2抑制剂处理此类模型会导致磷酸化ERK1/2(p-ERK)水平最初下降,然而p-ERK水平在两小时内迅速反弹。这种p-ERK反弹被FGFR抑制剂或高剂量的SHP2抑制剂阻断。从机制上讲,与由EGFR驱动的细胞相比,由FGFR驱动的细胞倾向于表达高水平的RTK负调节因子,如SPRY家族蛋白,这些蛋白在ERK抑制后会迅速下调。此外,在由FGFR驱动的细胞中过表达SPRY4可防止MAPK途径重新激活,并使它们对SHP2抑制剂敏感。我们还确定了两种新的联合方法来提高SHP2抑制剂的疗效,一种是与不同位点2的变构SHP2抑制剂联合,另一种是与RAS-SOS1相互作用抑制剂联合。我们的研究结果表明,SHP2抑制剂最初抑制途径后FGFR的快速反馈激活可能会促进SHP2的开放构象并导致对SHP2抑制剂产生抗性。这些发现可能有助于在SHP2抑制剂的临床开发中优化患者选择并预测抗性机制,并为发现对上游反馈激活更有效的SHP2抑制剂提供策略。