Department of Ophthalmology and Visual Science, Havener Eye Institute, The Ohio State University, Columbus, Ohio; Division of Human Genetics, Department of Internal Medicine, The Ohio State University, Columbus, Ohio.
Department of Ophthalmology and Visual Science, Havener Eye Institute, The Ohio State University, Columbus, Ohio.
Ophthalmology. 2020 May;127(5):668-678. doi: 10.1016/j.ophtha.2019.11.009. Epub 2019 Nov 18.
To identify susceptibility genes associated with hereditary predisposition to uveal melanoma (UM) in patients with no detectable germline BAP1 alterations.
Retrospective case series from academic referral centers.
Cohort of 154 UM patients with high risk of hereditary cancer defined as patients with 1 or more of the following: (1) familial UM, (2) young age (<35 years) at diagnosis, (3) personal history of other primary cancers, and (4) family history of 2 or more primary cancers with no detectable mutation or deletion in BAP1 gene.
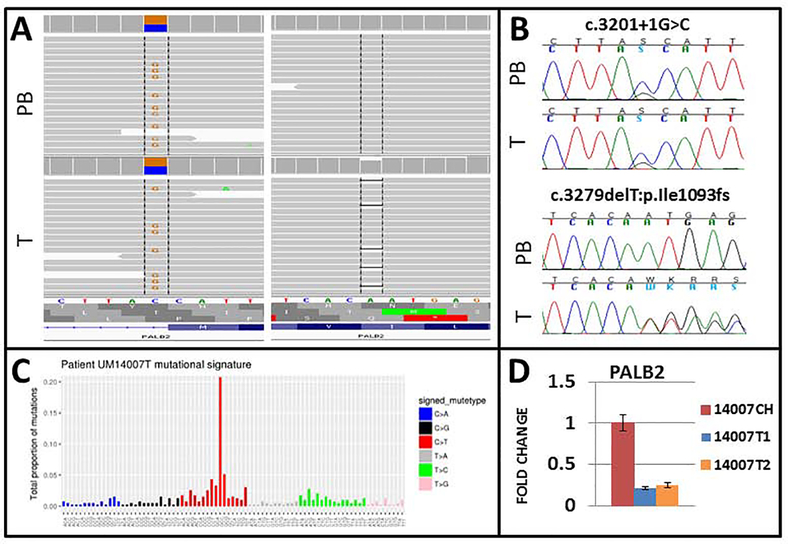
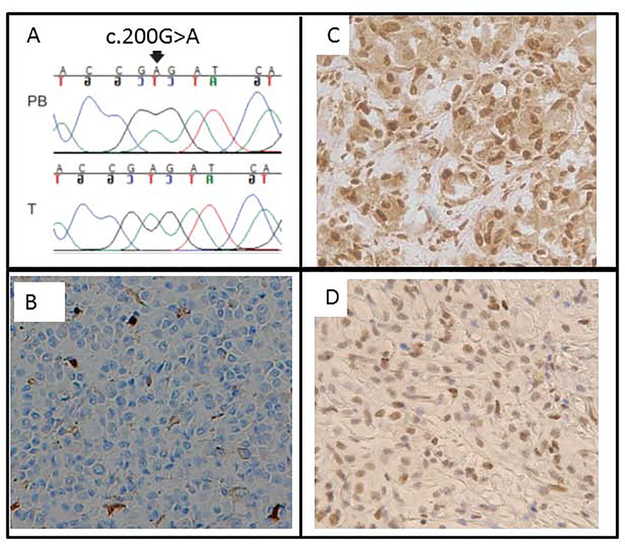
Whole exome sequencing, a cancer gene panel, or both were carried out. Probands included 27 patients with familial UM, 1 patient with bilateral UM, 1 patient with congenital UM, and 125 UM patients with strong personal or family histories, or both, of cancer. Functional validation of variants was carried out by immunohistochemistry, reverse-transcriptase polymerase chain reaction, and genotyping.
Clinical characterization of UM patients with germline alterations in known cancer genes.
We identified actionable pathogenic variants in 8 known hereditary cancer predisposition genes (PALB2, MLH1, MSH6, CHEK2, SMARCE1, ATM, BRCA1, and CTNNA1) in 9 patients, including 3 of 27 patients (11%) with familial UM and 6 of 127 patients (4.7%) with a high risk for cancer. Two patients showed pathogenic variants in CHEK2 and PALB2, whereas variants in the other genes each occurred in 1 patient. Biallelic inactivation of PALB2 and MLH1 was observed in tumors from the respective patients. The frequencies of pathogenic variants in PALB2, MLH1, and SMARCE1 in UM patients were significantly higher than the observed frequencies in noncancer controls (PALB2: P = 0.02; odds ratio, 8.9; 95% confidence interval, 1.5-30.6; MLH1: P = 0.04; odds ratio, 25.4; 95% confidence interval, 1.2-143; SMARCE1: P = 0.001; odds ratio, 2047; 95% confidence interval, 52-4.5e15, respectively).
The study provided moderate evidence of gene and disease association of germline mutations in PALB2 and MLH1 with hereditary predisposition to UM. It also identified several other candidate susceptibility genes. The results suggest locus heterogeneity in predisposition to UM. Genetic testing for hereditary predisposition to cancer is warranted in UM patients with strong personal or family history of cancers, or both.
鉴定与无胚系 BAP1 改变的葡萄膜黑色素瘤(UM)患者遗传易感性相关的易感基因。
来自学术转诊中心的回顾性病例系列。
队列包括 154 名 UM 高危遗传性癌症患者,高危定义为以下 1 种或多种情况:(1)家族性 UM;(2)诊断时年龄较小(<35 岁);(3)有其他原发性癌症病史;(4)有 2 个或多个原发性癌症家族史,且 BAP1 基因无检测到的突变或缺失。
进行全外显子组测序、癌症基因panel 或两者兼用。先证者包括 27 名家族性 UM 患者、1 名双侧 UM 患者、1 名先天性 UM 患者和 125 名 UM 患者,他们有强烈的个人或家族癌症史,或两者兼有。通过免疫组织化学、逆转录酶聚合酶链反应和基因分型对变体进行功能验证。
已知癌症基因胚系改变的 UM 患者的临床特征。
我们在 9 名患者的 8 个已知遗传性癌症易感性基因(PALB2、MLH1、MSH6、CHEK2、SMARCE1、ATM、BRCA1 和 CTNNA1)中发现了可操作的致病性变异,包括 3 名家族性 UM 患者(11%)和 6 名癌症高危患者(4.7%)。2 名患者存在 CHEK2 和 PALB2 的致病性变异,而其他基因的每个患者均存在 1 个致病性变异。在各自患者的肿瘤中观察到 PALB2 和 MLH1 的双等位基因失活。PALB2、MLH1 和 SMARCE1 中致病性变异在 UM 患者中的频率明显高于非癌症对照(PALB2:P=0.02;优势比,8.9;95%置信区间,1.5-30.6;MLH1:P=0.04;优势比,25.4;95%置信区间,1.2-143;SMARCE1:P=0.001;优势比,2047;95%置信区间,52-4.5e15)。
该研究提供了中等证据,表明胚系 PALB2 和 MLH1 突变与 UM 的遗传易感性相关。它还确定了其他几个候选易感基因。结果表明 UM 易感性存在基因座异质性。在 UM 患者中有强烈的个人或家族癌症史,或两者兼有时,应进行遗传性癌症易感性检测。