Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, USA.
Department of Pediatrics, Division of Medical Genetics, Duke University Medical Center, Durham, North Carolina, USA.
Am J Med Genet A. 2020 May;182(5):1053-1065. doi: 10.1002/ajmg.a.61518. Epub 2020 Feb 21.
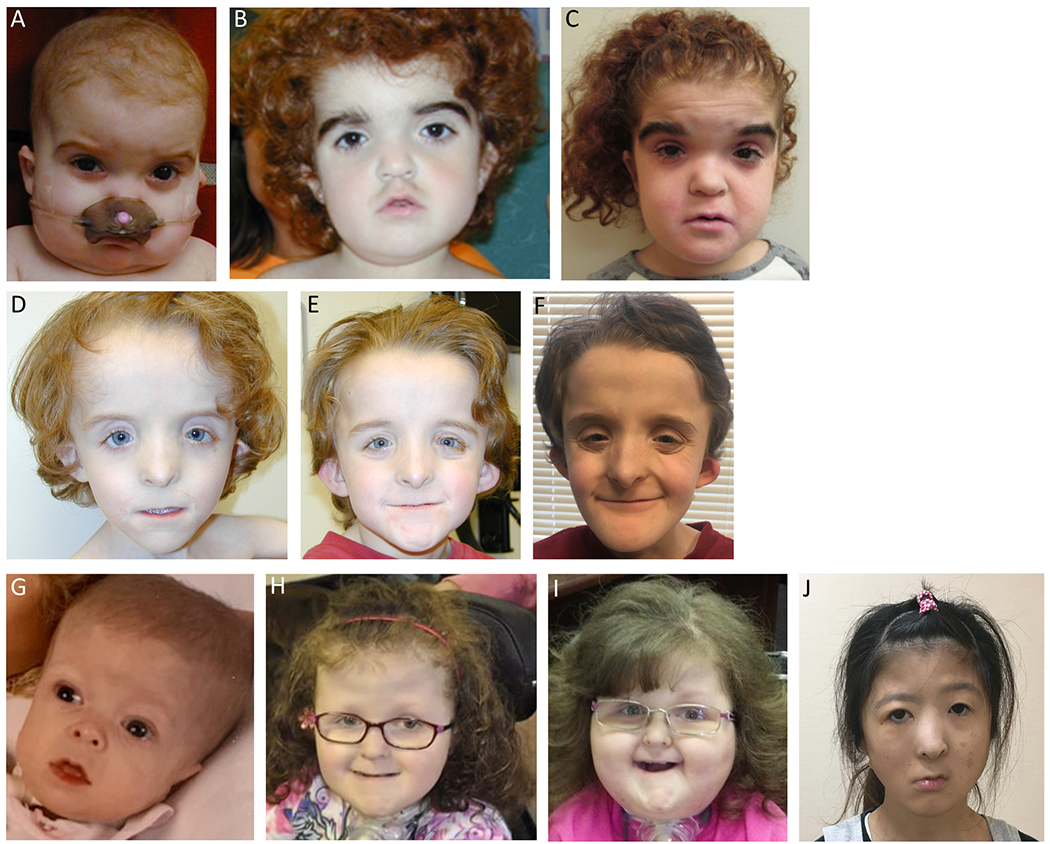
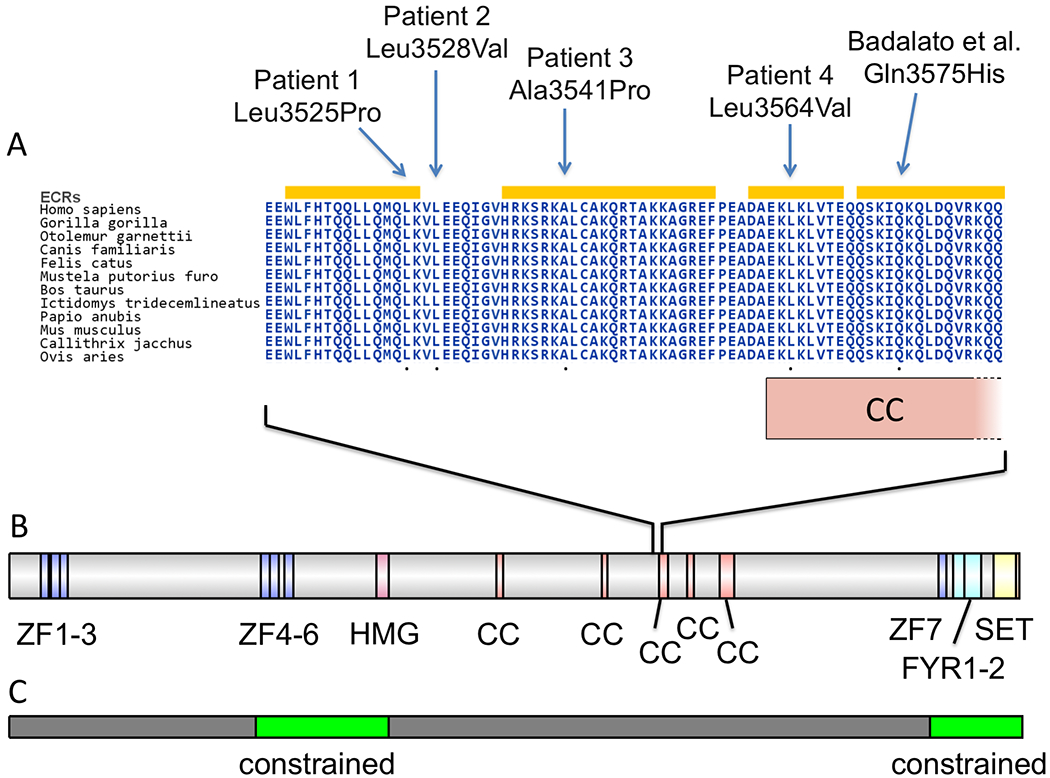
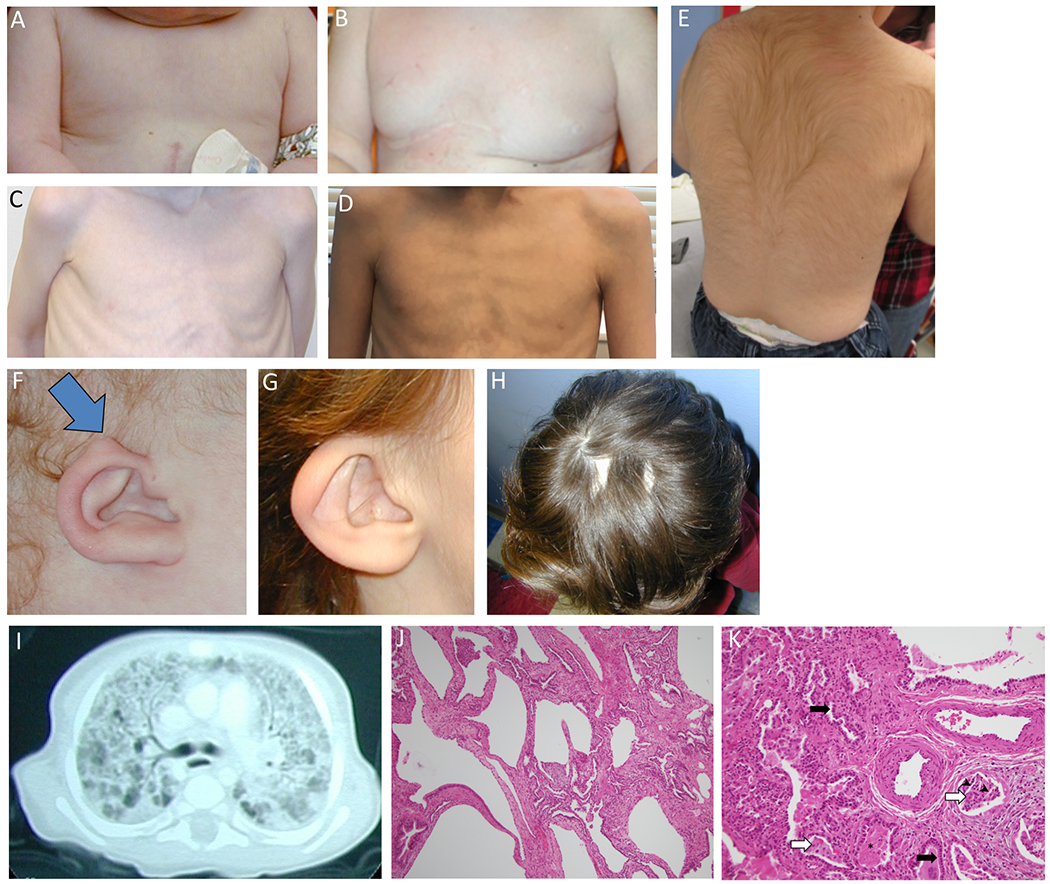
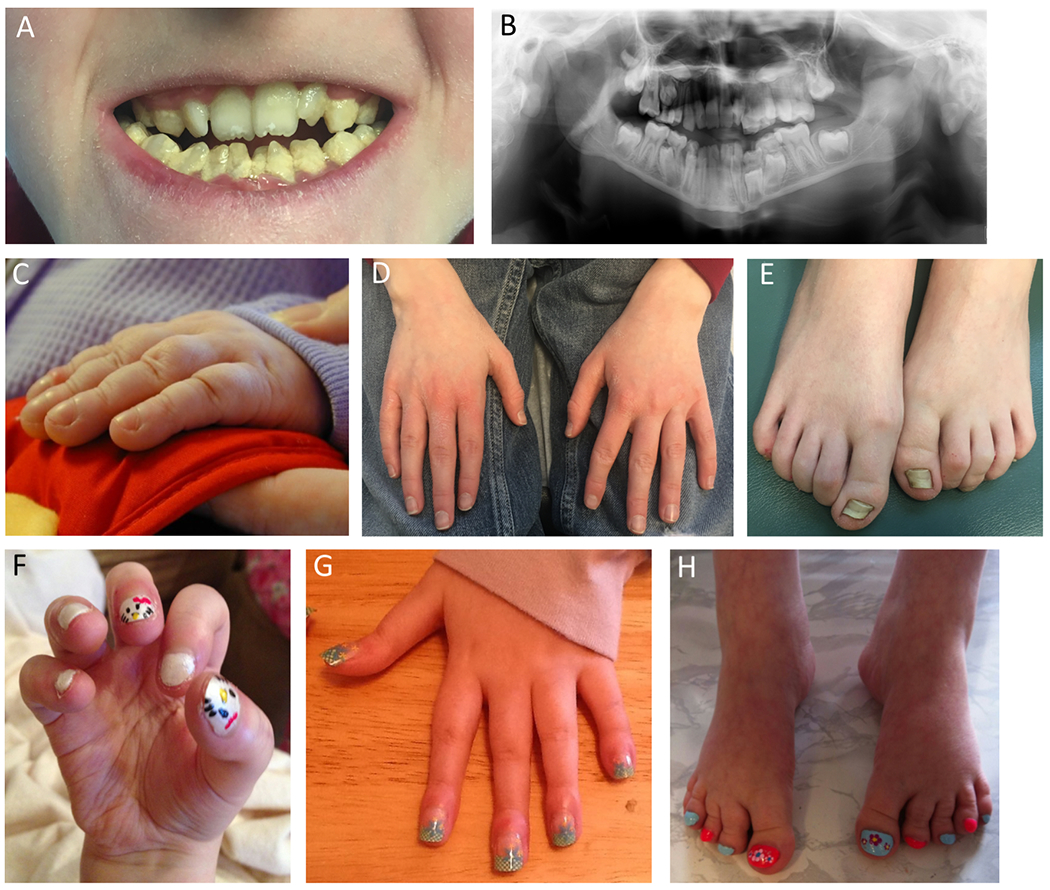
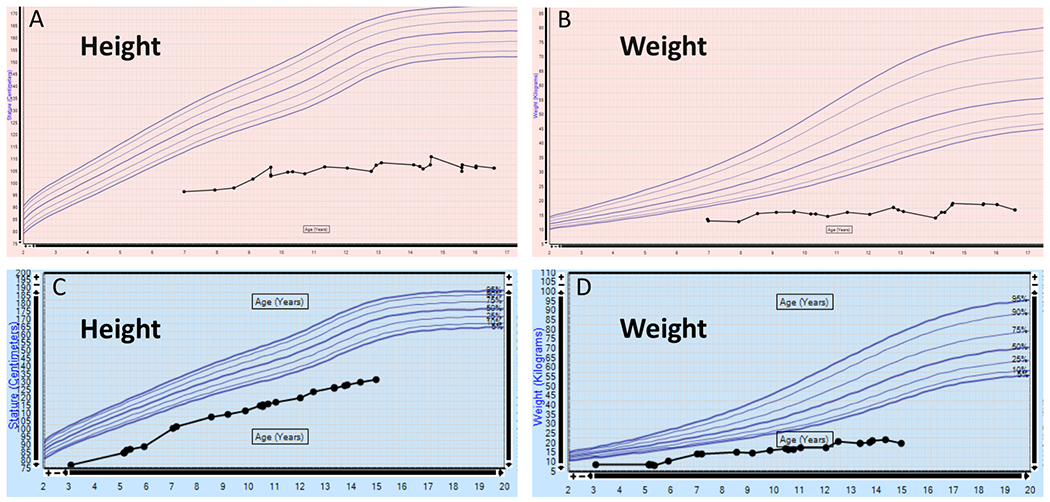
Pathogenic variants in KMT2D, which encodes lysine specific methyltransferase 2D, cause autosomal dominant Kabuki syndrome, associated with distinctive dysmorphic features including arched eyebrows, long palpebral fissures with eversion of the lower lid, large protuberant ears, and fetal finger pads. Most disease-causing variants identified to date are putative loss-of-function alleles, although 15-20% of cases are attributed to missense variants. We describe here four patients (including one previously published patient) with de novo KMT2D missense variants and with shared but unusual clinical findings not typically seen in Kabuki syndrome, including athelia (absent nipples), choanal atresia, hypoparathyroidism, delayed or absent pubertal development, and extreme short stature. These individuals also lack the typical dysmorphic facial features found in Kabuki syndrome. Two of the four patients had severe interstitial lung disease. All of these variants cluster within a 40-amino-acid region of the protein that is located just N-terminal of an annotated coiled coil domain. These findings significantly expand the phenotypic spectrum of features associated with variants in KMT2D beyond those seen in Kabuki syndrome and suggest a possible new underlying disease mechanism for these patients.
致病性变异在 KMT2D 中,其编码赖氨酸特异性甲基转移酶 2D,导致常染色体显性遗传的歌舞伎综合征,伴有独特的发育异常特征,包括拱形眉毛、上睑下垂的长睑裂、大而突出的耳朵和胎儿指垫。迄今为止,已鉴定出的大多数致病变异是假定的功能丧失等位基因,尽管 15-20%的病例归因于错义变异。我们在这里描述了四个患有 KMT2D 错义变异的新生患者(包括一个以前发表的患者),这些患者具有共同但不常见的临床发现,这些发现通常不会在歌舞伎综合征中看到,包括无乳头(athelia)、后鼻孔闭锁、甲状旁腺功能减退、青春期发育延迟或缺失,以及极端身材矮小。这些个体也缺乏在歌舞伎综合征中发现的典型面部发育异常特征。其中两个患者患有严重的间质性肺病。所有这些变异都聚集在蛋白的 40 个氨基酸区域内,该区域位于注释的卷曲螺旋域的 N 端附近。这些发现显著扩展了与 KMT2D 变异相关的表型谱,超出了在歌舞伎综合征中看到的特征,并提示这些患者可能存在新的潜在疾病机制。