Laboratory of Genome Maintenance, The Rockefeller University, New York, NY.
Cancer Genomics Unit, Cancer Genetics and Comparative Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD.
Blood. 2020 Apr 30;135(18):1588-1602. doi: 10.1182/blood.2019003249.
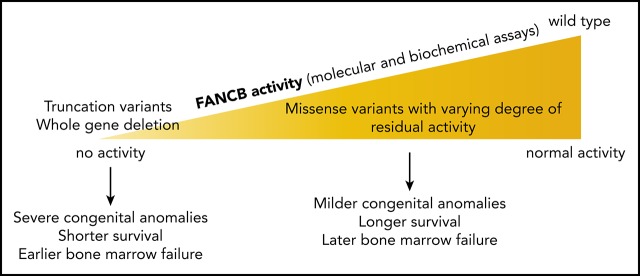
Fanconi anemia (FA) is the most common genetic cause of bone marrow failure and is caused by inherited pathogenic variants in any of 22 genes. Of these, only FANCB is X-linked. We describe a cohort of 19 children with FANCB variants, from 16 families of the International Fanconi Anemia Registry. Those with FANCB deletion or truncation demonstrate earlier-than-average onset of bone marrow failure and more severe congenital abnormalities compared with a large series of FA individuals in published reports. This reflects the indispensable role of FANCB protein in the enzymatic activation of FANCD2 monoubiquitination, an essential step in the repair of DNA interstrand crosslinks. For FANCB missense variants, more variable severity is associated with the extent of residual FANCD2 monoubiquitination activity. We used transcript analysis, genetic complementation, and biochemical reconstitution of FANCD2 monoubiquitination to determine the pathogenicity of each variant. Aberrant splicing and transcript destabilization were associated with 2 missense variants. Individuals carrying missense variants with drastically reduced FANCD2 monoubiquitination in biochemical and/or cell-based assays tended to show earlier onset of hematologic disease and shorter survival. Conversely, variants with near-normal FANCD2 monoubiquitination were associated with more favorable outcome. Our study reveals a genotype-phenotype correlation within the FA-B complementation group of FA, where severity is associated with level of residual FANCD2 monoubiquitination.
范可尼贫血症(FA)是最常见的遗传性骨髓衰竭症,由 22 个基因中的任何一个遗传致病性变异引起。其中,只有 FANCB 是 X 连锁的。我们描述了来自国际范可尼贫血症登记处的 16 个家庭的 19 名 FANCB 变异儿童的队列。与已发表报告中的大量 FA 个体相比,那些具有 FANCB 缺失或截断的个体表现出早于平均发病的骨髓衰竭和更严重的先天性异常。这反映了 FANCB 蛋白在 FANCD2 单泛素化酶激活中的不可或缺作用,这是修复 DNA 链间交联的关键步骤。对于 FANCB 错义变异,与剩余 FANCD2 单泛素化活性的程度相关的是更可变的严重程度。我们使用转录分析、遗传互补和 FANCD2 单泛素化的生化重建来确定每个变异的致病性。异常剪接和转录失稳与 2 个错义变异相关。在生化和/或基于细胞的测定中具有 FANCD2 单泛素化明显减少的错义变异个体往往表现出更早发病的血液疾病和更短的生存期。相反,具有接近正常 FANCD2 单泛素化的变异与更好的预后相关。我们的研究揭示了 FA-B 互补组内 FA 的基因型-表型相关性,其中严重程度与残留 FANCD2 单泛素化的水平相关。