Institute of Immunity and Transplantation, Division of Infection and Immunity, University College London, London, United Kingdom.
Institute of Structural and Molecular Biology, Birkbeck College, London, United Kingdom.
PLoS Comput Biol. 2020 Feb 28;16(2):e1007710. doi: 10.1371/journal.pcbi.1007710. eCollection 2020 Feb.
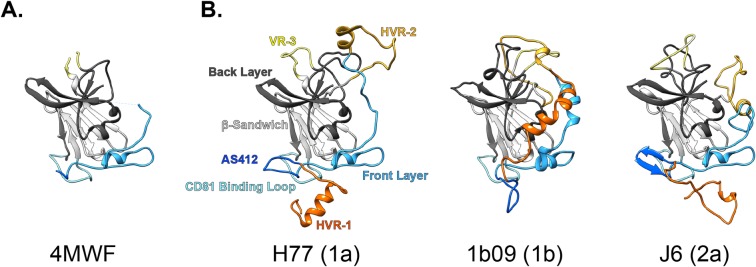
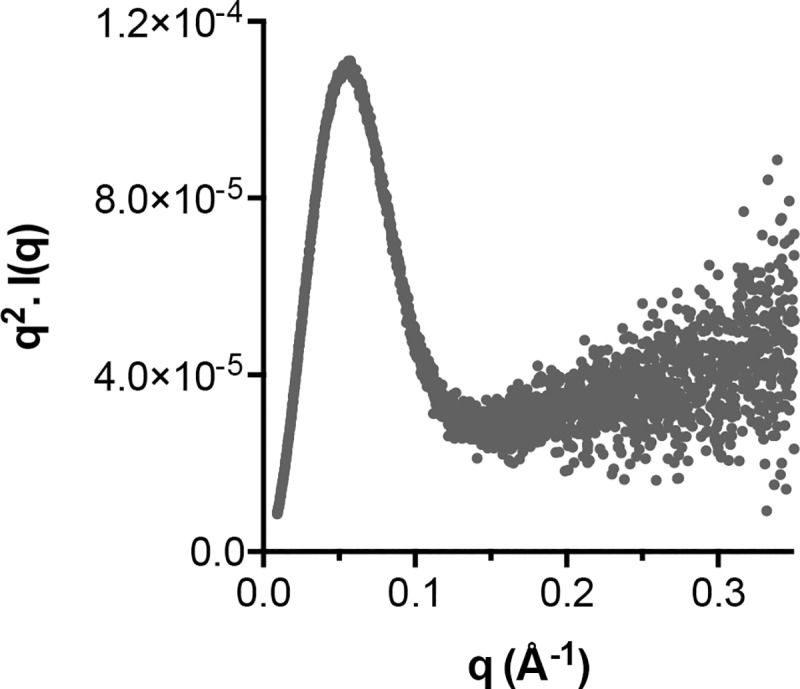
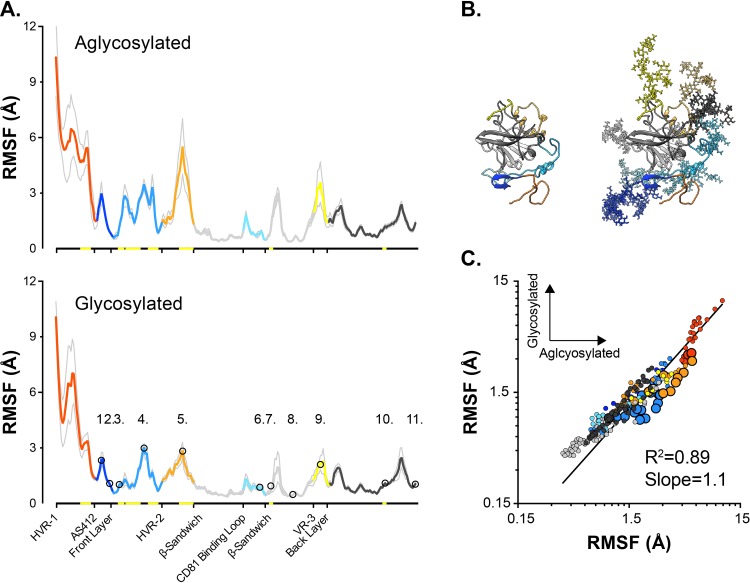
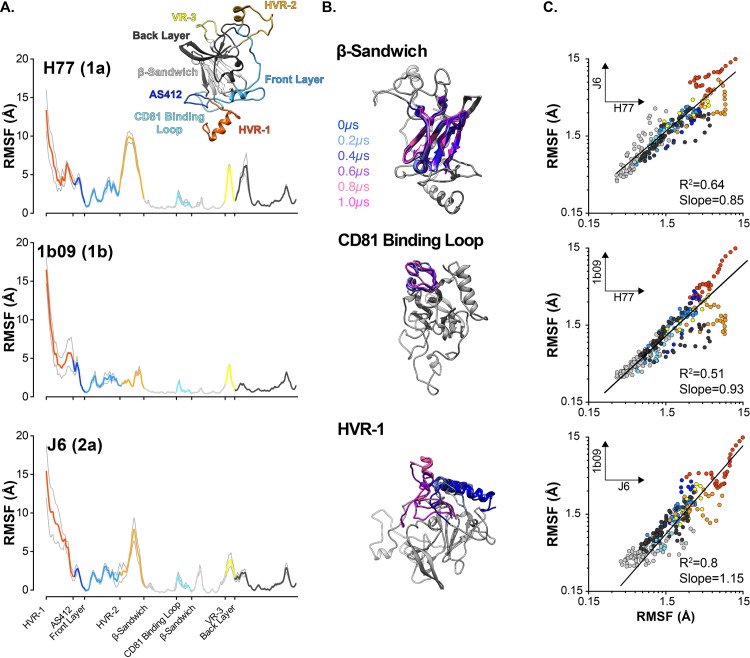
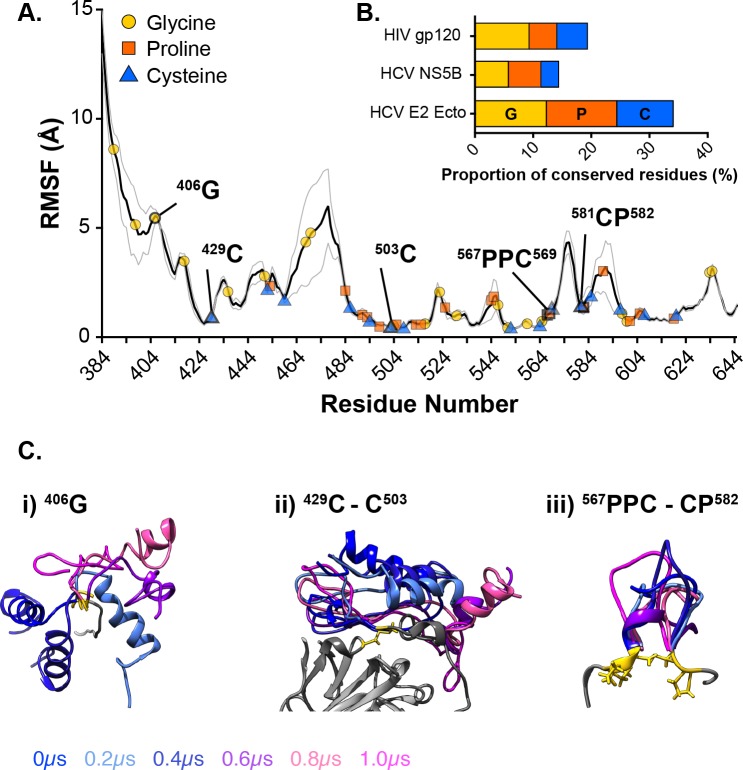
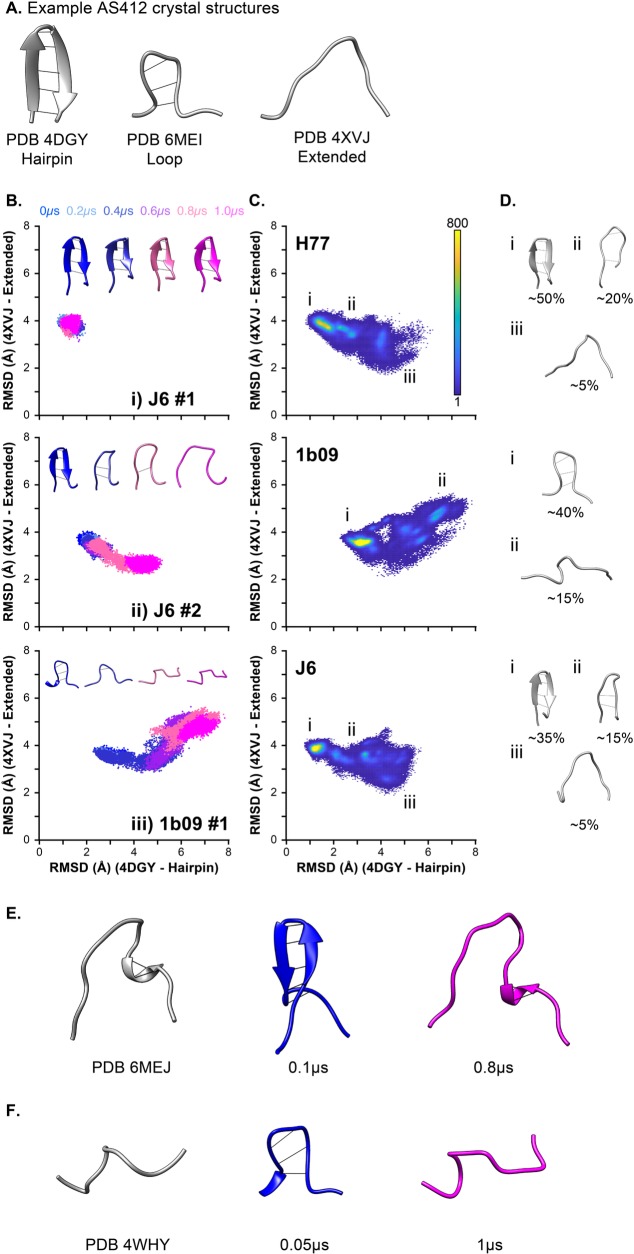
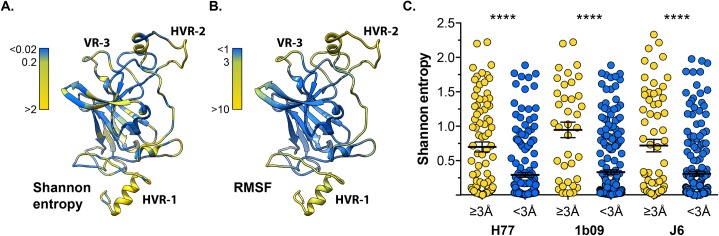
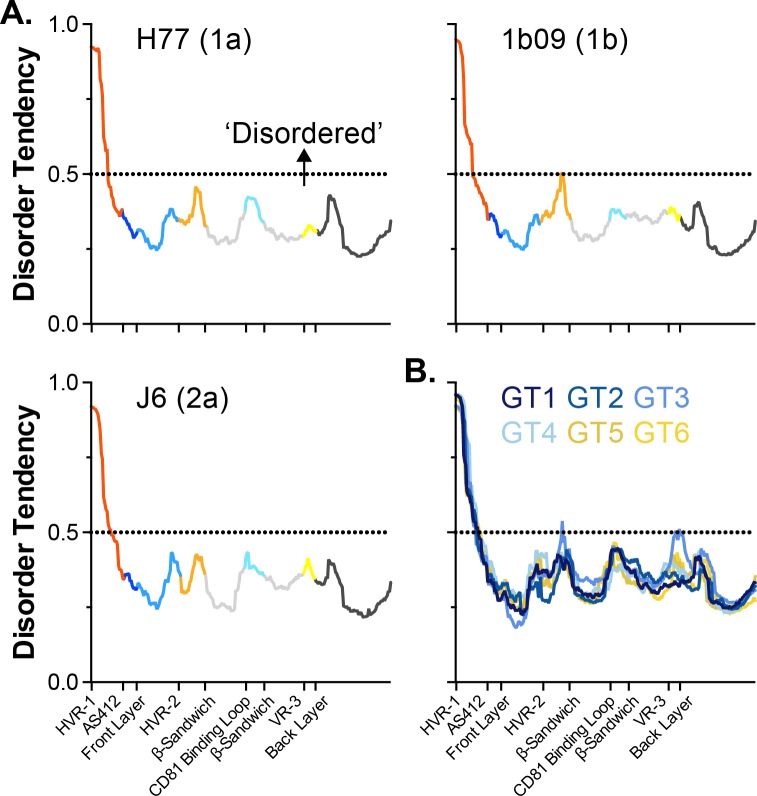
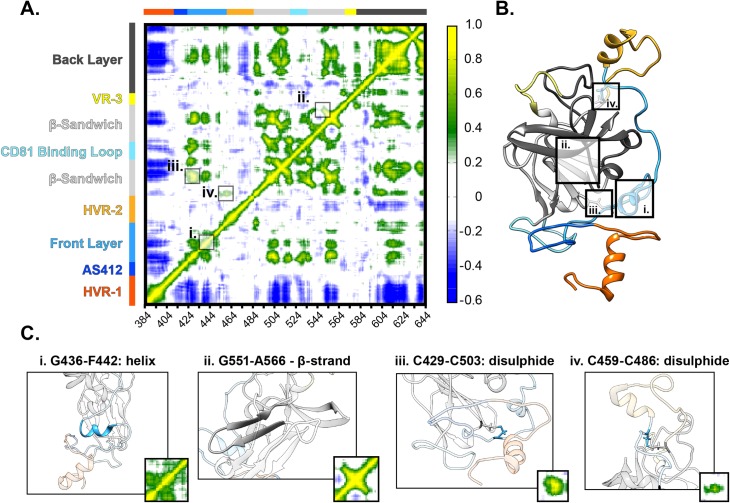
The glycoproteins of hepatitis C virus, E1E2, are unlike any other viral fusion machinery yet described, and are the current focus of immunogen design in HCV vaccine development; thus, making E1E2 both scientifically and medically important. We used pre-existing, but fragmentary, structures to model a complete ectodomain of the major glycoprotein E2 from three strains of HCV. We then performed molecular dynamic simulations to explore the conformational landscape of E2, revealing a number of important features. Despite high sequence divergence, and subtle differences in the models, E2 from different strains behave similarly, possessing a stable core flanked by highly flexible regions, some of which perform essential functions such as receptor binding. Comparison with sequence data suggest that this consistent behaviour is conferred by a network of conserved residues that act as hinge and anchor points throughout E2. The variable regions (HVR-1, HVR-2 and VR-3) exhibit particularly high flexibility, and bioinformatic analysis suggests that HVR-1 is a putative intrinsically disordered protein region. Dynamic cross-correlation analyses demonstrate intramolecular communication and suggest that specific regions, such as HVR-1, can exert influence throughout E2. To support our computational approach we performed small-angle X-ray scattering with purified E2 ectodomain; this data was consistent with our MD experiments, suggesting a compact globular core with peripheral flexible regions. This work captures the dynamic behaviour of E2 and has direct relevance to the interaction of HCV with cell-surface receptors and neutralising antibodies.
丙型肝炎病毒的糖蛋白 E1E2 与任何其他病毒融合机制都不同,是当前丙型肝炎病毒疫苗开发中免疫原设计的重点;因此,E1E2 既具有科学意义,也具有医学意义。我们使用现有的但不完整的结构来模拟丙型肝炎病毒三种毒株主要糖蛋白 E2 的完整外域。然后,我们进行了分子动力学模拟,以探索 E2 的构象景观,揭示了一些重要的特征。尽管存在高度的序列差异和模型中的细微差异,但来自不同毒株的 E2 表现相似,具有稳定的核心,周围是高度灵活的区域,其中一些区域执行诸如受体结合等基本功能。与序列数据的比较表明,这种一致的行为是由保守残基网络赋予的,这些残基在整个 E2 中充当铰链和锚固点。可变区(HVR-1、HVR-2 和 VR-3)表现出特别高的灵活性,生物信息学分析表明 HVR-1 是一个潜在的无序蛋白区域。动态互相关分析表明分子内存在通讯,并表明特定区域,如 HVR-1,可以在整个 E2 中发挥影响。为了支持我们的计算方法,我们对纯化的 E2 外域进行了小角度 X 射线散射;该数据与我们的 MD 实验一致,表明存在一个紧凑的球形核心和外围灵活的区域。这项工作捕获了 E2 的动态行为,与丙型肝炎病毒与细胞表面受体和中和抗体的相互作用直接相关。