Wang Chunyu, Zhang Jie, Wang Xueping, Han Ke, Guo Maozu
School of Computer Science and Technology, Harbin Institute of Technology, Harbin, China.
School of Computer and Information Engineering, Harbin University of Commerce, Harbin, China.
Front Genet. 2020 Feb 4;11:5. doi: 10.3389/fgene.2020.00005. eCollection 2020.
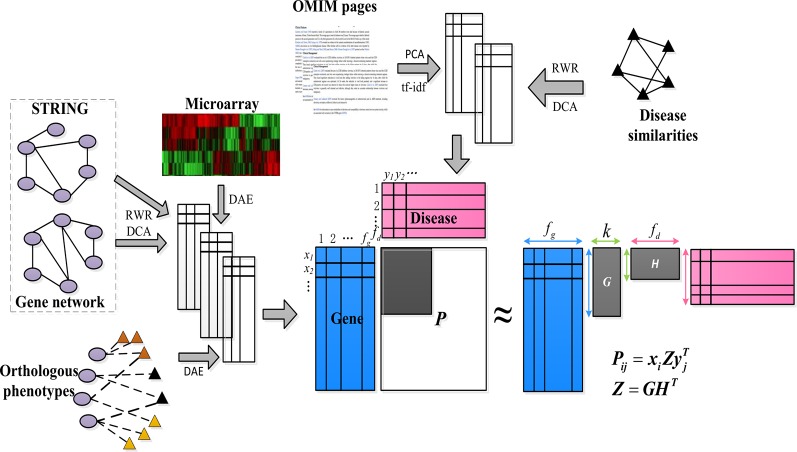
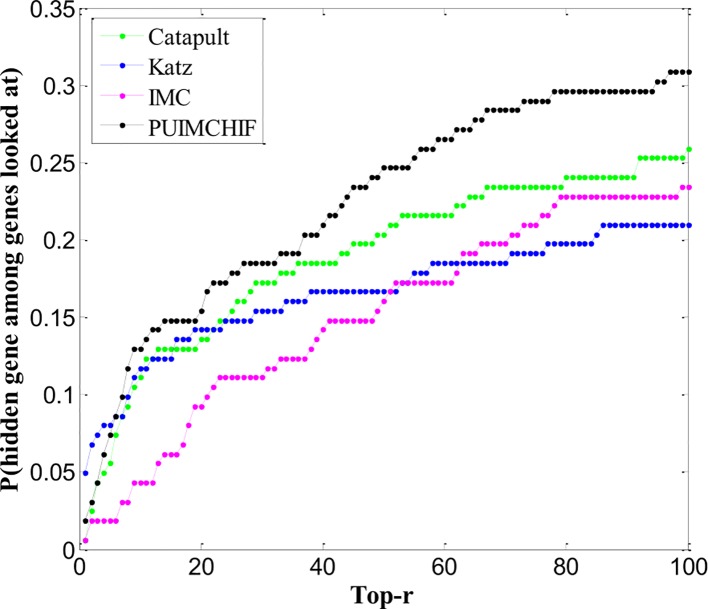
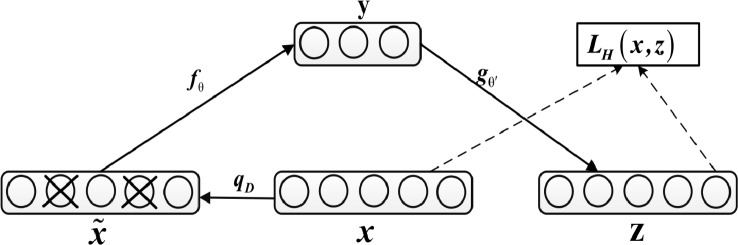
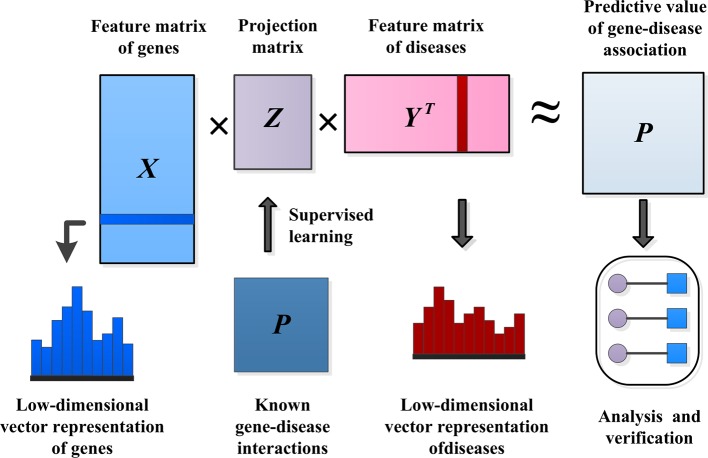
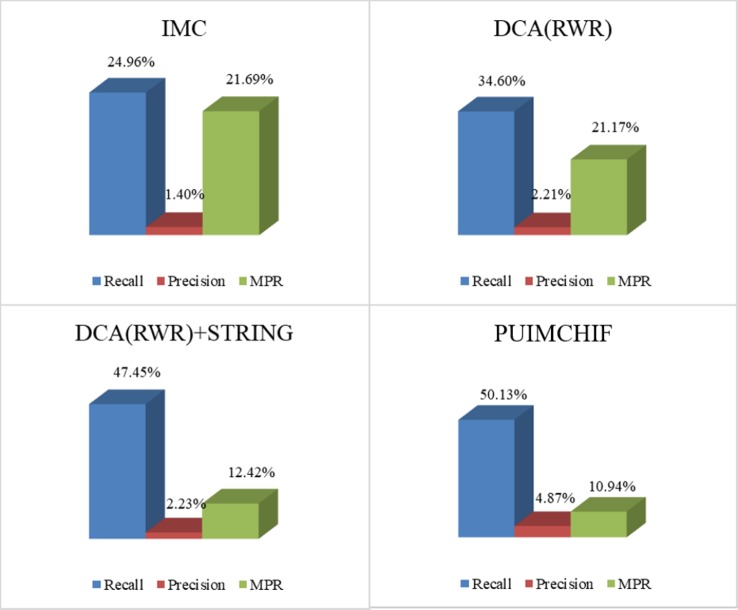
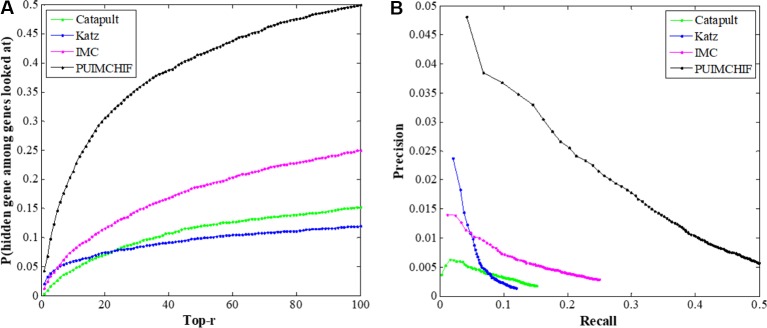
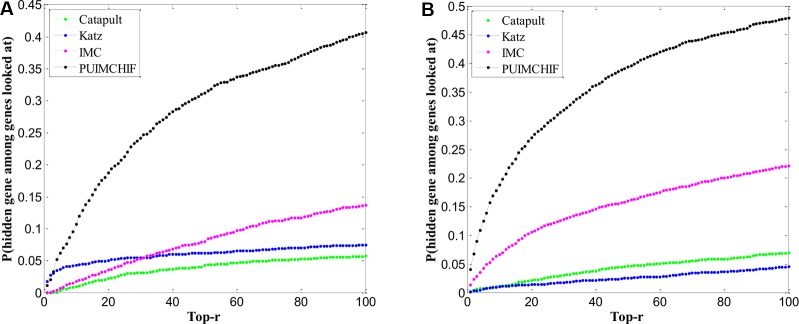
Complex diseases seriously affect people's physical and mental health. The discovery of disease-causing genes has become a target of research. With the emergence of bioinformatics and the rapid development of biotechnology, to overcome the inherent difficulties of the long experimental period and high cost of traditional biomedical methods, researchers have proposed many gene prioritization algorithms that use a large amount of biological data to mine pathogenic genes. However, because the currently known gene-disease association matrix is still very sparse and lacks evidence that genes and diseases are unrelated, there are limits to the predictive performance of gene prioritization algorithms. Based on the hypothesis that functionally related gene mutations may lead to similar disease phenotypes, this paper proposes a PU induction matrix completion algorithm based on heterogeneous information fusion (PUIMCHIF) to predict candidate genes involved in the pathogenicity of human diseases. On the one hand, PUIMCHIF uses different compact feature learning methods to extract features of genes and diseases from multiple data sources, making up for the lack of sparse data. On the other hand, based on the prior knowledge that most of the unknown gene-disease associations are unrelated, we use the PU-Learning strategy to treat the unknown unlabeled data as negative examples for biased learning. The experimental results of the PUIMCHIF algorithm regarding the three indexes of precision, recall, and mean percentile ranking (MPR) were significantly better than those of other algorithms. In the top 100 global prediction analysis of multiple genes and multiple diseases, the probability of recovering true gene associations using PUIMCHIF reached 50% and the MPR value was 10.94%. The PUIMCHIF algorithm has higher priority than those from other methods, such as IMC and CATAPULT.
复杂疾病严重影响人们的身心健康。致病基因的发现已成为研究目标。随着生物信息学的出现和生物技术的快速发展,为克服传统生物医学方法实验周期长、成本高的固有困难,研究人员提出了许多基因优先级排序算法,这些算法利用大量生物数据挖掘致病基因。然而,由于目前已知的基因-疾病关联矩阵仍然非常稀疏,且缺乏基因与疾病不相关的证据,基因优先级排序算法的预测性能存在局限性。基于功能相关基因突变可能导致相似疾病表型的假设,本文提出了一种基于异构信息融合的PU诱导矩阵补全算法(PUIMCHIF),用于预测参与人类疾病致病性的候选基因。一方面,PUIMCHIF使用不同的紧凑特征学习方法从多个数据源提取基因和疾病的特征,弥补稀疏数据的不足。另一方面,基于大多数未知基因-疾病关联不相关的先验知识,我们使用PU学习策略将未知的未标记数据作为负例进行有偏学习。PUIMCHIF算法在精确率、召回率和平均百分位排名(MPR)这三个指标上的实验结果明显优于其他算法。在多个基因和多种疾病的前100名全局预测分析中,使用PUIMCHIF恢复真实基因关联的概率达到50%,MPR值为10.94%。PUIMCHIF算法比其他方法(如IMC和CATAPULT)具有更高的优先级。