Department of Molecular and Systems Biology, Geisel School of Medicine at Dartmouth, Hanover, NH, 03755, USA.
Department of Obstetrics & Gynecology, NYC Health + Hospitals/Coney Island, Brooklyn, NY, 11235, USA.
Lab Invest. 2020 Oct;100(10):1356-1366. doi: 10.1038/s41374-020-0413-8. Epub 2020 Mar 6.
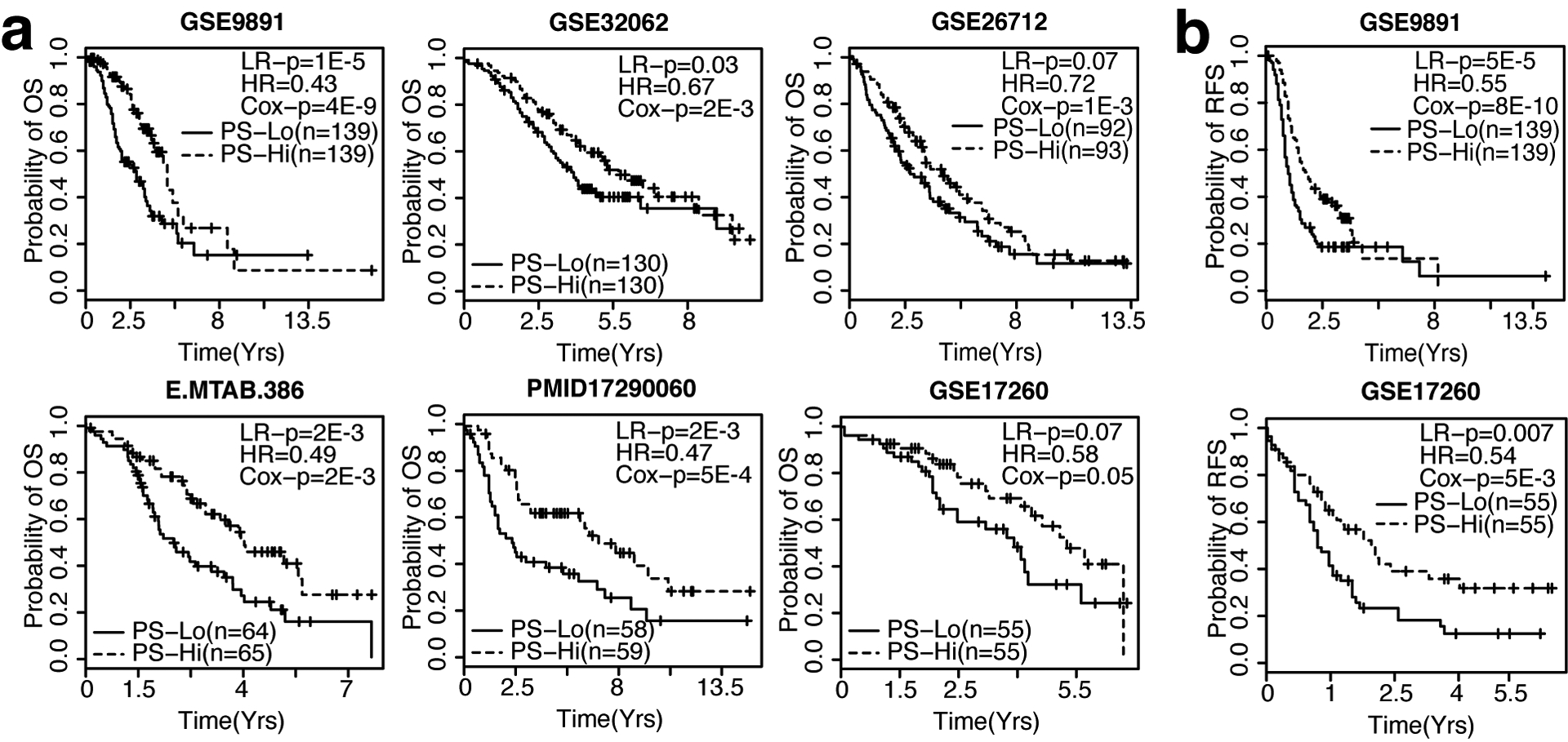
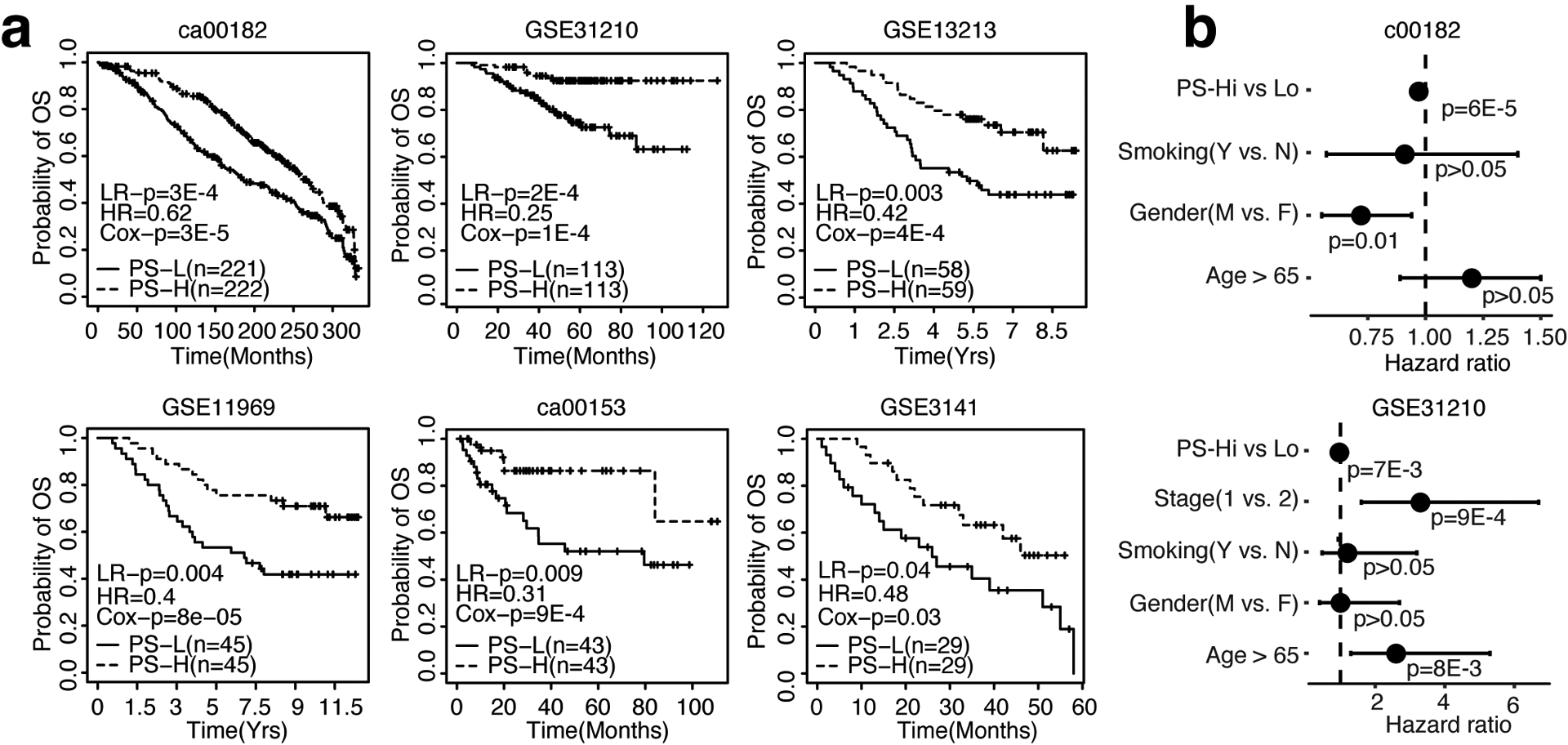
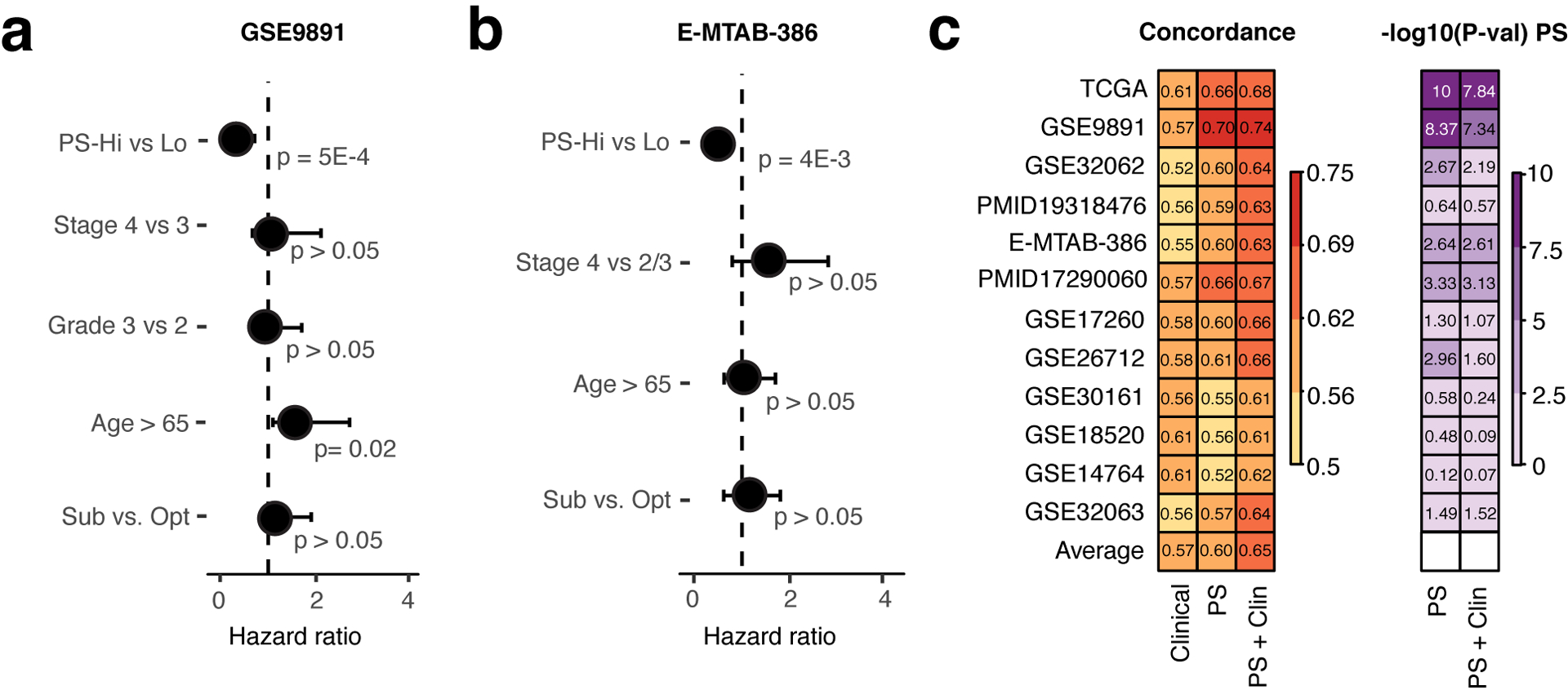
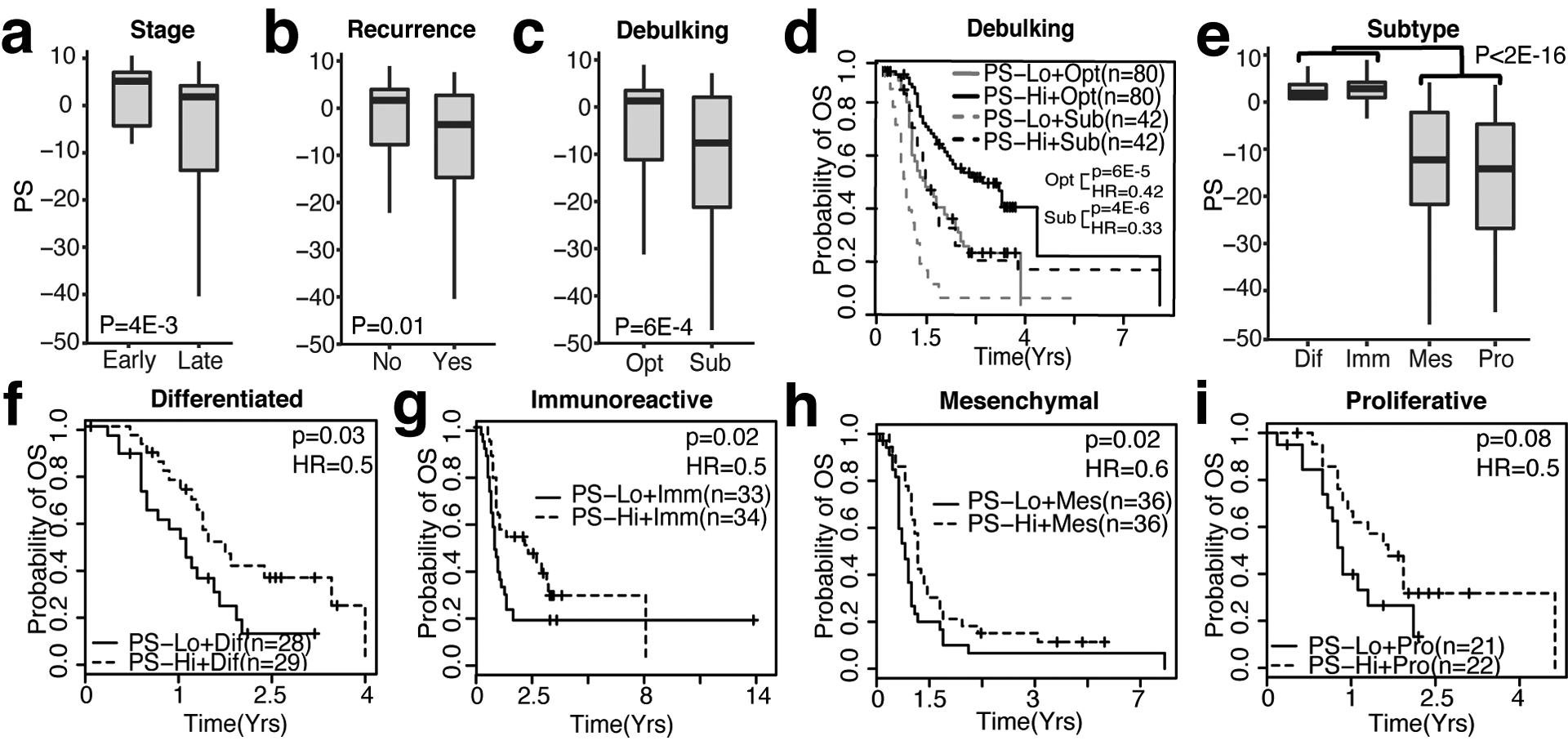
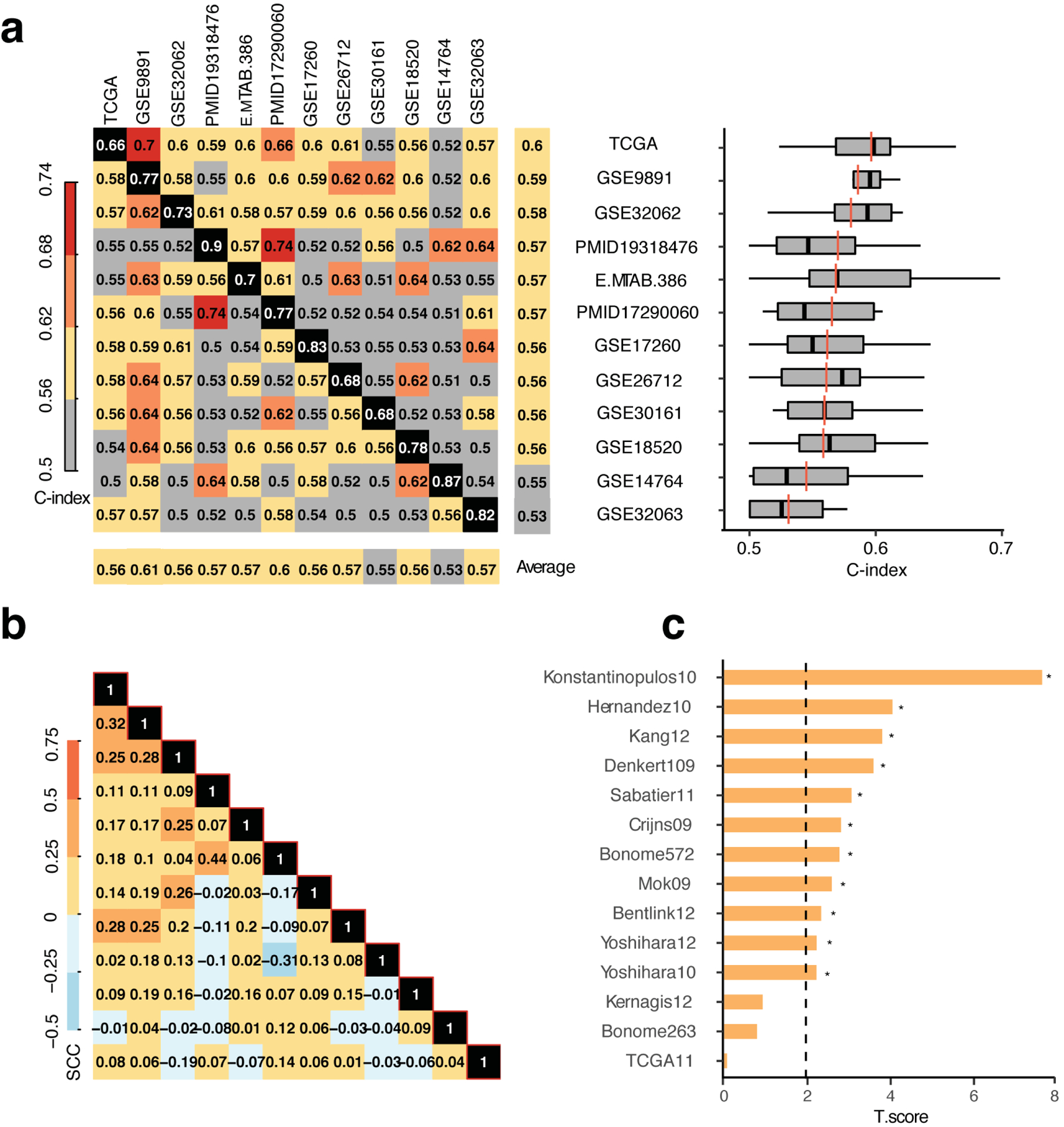
Developing prognostic biomarkers for specific cancer types that accurately predict patient survival is increasingly important in clinical research and practice. Despite the enormous potential of prognostic signatures, proposed models have found limited implementations in routine clinical practice. Herein, we propose a generic, RNA sequencing platform independent, statistical framework named whole transcriptome signature for prognostic prediction to generate prognostic gene signatures. Using ovarian cancer and lung adenocarcinoma as examples, we provide evidence that our prognostic signatures overperform previous reported signatures, capture prognostic features not explained by clinical variables, and expose biologically relevant prognostic pathways, including those involved in the immune system and cell cycle. Our approach demonstrates a robust method for developing prognostic gene expression signatures. In conclusion, our statistical framework can be generally applied to all cancer types for prognostic prediction and might be extended to other human diseases. The proposed method is implemented as an R package (PanCancerSig) and is freely available on GitHub ( https://github.com/Cheng-Lab-GitHub/PanCancer_Signature ).
开发针对特定癌症类型的预后生物标志物,以准确预测患者的生存率,在临床研究和实践中变得越来越重要。尽管预后标志物具有巨大的潜力,但提出的模型在常规临床实践中的应用有限。在此,我们提出了一个通用的、不受 RNA 测序平台影响的、名为全转录组signature for prognostic prediction 的统计框架,用于生成预后基因标志物。我们以卵巢癌和肺腺癌为例,提供了证据表明,我们的预后标志物优于以前报道的标志物,能够捕获临床变量无法解释的预后特征,并揭示了生物学上相关的预后途径,包括涉及免疫系统和细胞周期的途径。我们的方法为开发预后基因表达标志物提供了一种稳健的方法。总之,我们的统计框架可以广泛应用于所有癌症类型的预后预测,并且可以扩展到其他人类疾病。该方法作为一个 R 包(PanCancerSig)实现,并在 GitHub 上免费提供(https://github.com/Cheng-Lab-GitHub/PanCancer_Signature)。