Department of Physics and Astronomy, University of Bologna, Bologna, Italy.
Department of Computer Science and Engineering, University of Bologna, Bologna, Italy.
PLoS One. 2020 Mar 10;15(3):e0229763. doi: 10.1371/journal.pone.0229763. eCollection 2020.
Meta-analysis is a powerful means for leveraging the hundreds of experiments being run worldwide into more statistically powerful analyses. This is also true for the analysis of omic data, including genome-wide DNA methylation. In particular, thousands of DNA methylation profiles generated using the Illumina 450k are stored in the publicly accessible Gene Expression Omnibus (GEO) repository. Often, however, the intensity values produced by the BeadChip (raw data) are not deposited, therefore only pre-processed values -obtained after computational manipulation- are available. Pre-processing is possibly different among studies and may then affect meta-analysis by introducing non-biological sources of variability.
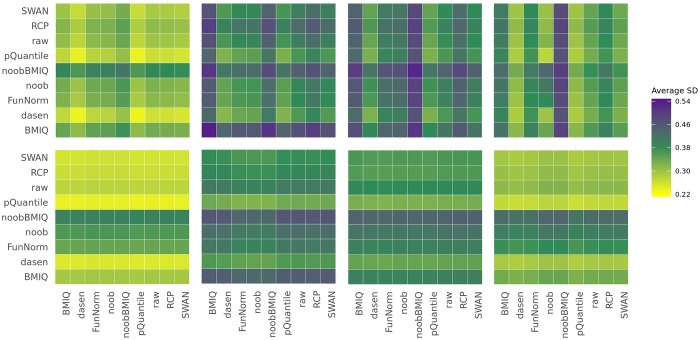
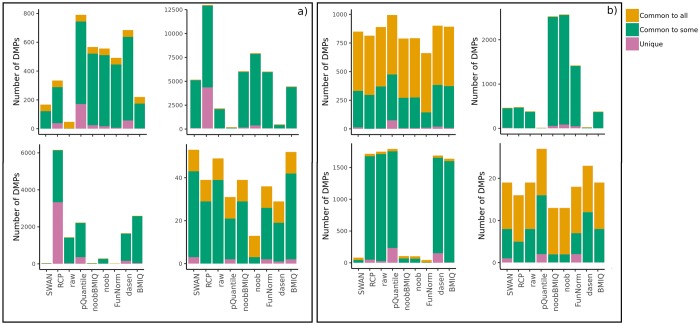
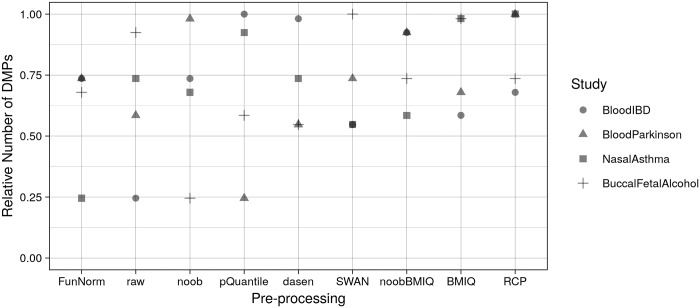
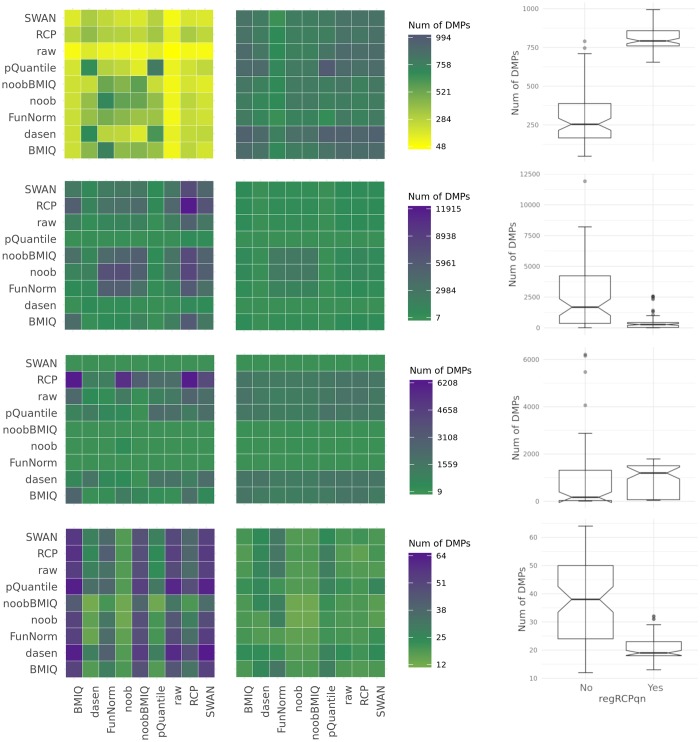
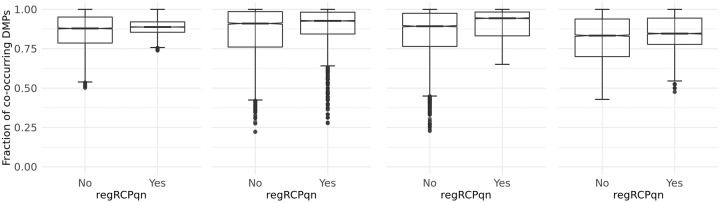
To systematically investigate the effect of pre-processing on meta-analysis, we analysed four different collections of DNA methylation samples (datasets), each composed of two subsets, for which raw data from controls (i.e. healthy subjects) and cases (i.e. patients) are available. We pre-processed the data from each dataset with nine among the most common pipelines found in literature. Moreover, we evaluated the performance of regRCPqn, a modification of the RCP algorithm that aims to improve data consistency. For each combination of pre-processing (9 × 9), we first evaluated the between-sample variability among control subjects and, then, we identified genomic positions that are differentially methylated between cases and controls (differential analysis).
The pre-processing of DNA methylation data affects both the between-sample variability and the loci identified as differentially methylated, and the effects of pre-processing are strongly dataset-dependent. By contrast, application of our renormalization algorithm regRCPqn: (i) reduces variability and (ii) increases agreement between meta-analysed datasets, both critical components of data harmonization.
元分析是一种强大的方法,可以利用全球范围内进行的数百项实验进行更具统计学意义的分析。这对于分析组学数据(包括全基因组 DNA 甲基化)也是如此。特别是,使用 Illumina 450k 生成的数千个 DNA 甲基化谱存储在可公开访问的基因表达综合数据库(GEO)存储库中。然而,通常情况下,BeadChip 产生的强度值(原始数据)并未被存储,因此仅可获得经过计算处理后的值-经过计算处理后获得的值。预处理可能因研究而异,因此可能会通过引入非生物学来源的变异性来影响元分析。
为了系统地研究预处理对元分析的影响,我们分析了四个不同的 DNA 甲基化样本(数据集),每个数据集由两个子集组成,每个子集都有对照(即健康受试者)和病例(即患者)的原始数据。我们使用文献中最常见的 9 种管道之一对每个数据集的数据进行预处理。此外,我们还评估了 regRCPqn 的性能,regRCPqn 是 RCP 算法的一种改进,旨在提高数据一致性。对于预处理的每种组合(9×9),我们首先评估对照个体之间的样本间变异性,然后确定病例和对照之间差异甲基化的基因组位置(差异分析)。
DNA 甲基化数据的预处理会影响样本间的变异性和被确定为差异甲基化的位置,并且预处理的效果强烈依赖于数据集。相比之下,我们的重新归一化算法 regRCPqn 的应用:(i)降低了变异性,(ii)增加了元分析数据集之间的一致性,这是数据协调的两个关键组成部分。