Systems Medicine of Metabolism and Signalling, Laboratory of Paediatrics, University of Groningen, University Medical Centre Groningen, The Netherlands.
Analytical Biosciences and Metabolomics, Division of Systems Biomedicine and Pharmacology, Leiden Academic Centre for Drug Research, Leiden University, The Netherlands.
FEBS J. 2020 Dec;287(23):5096-5113. doi: 10.1111/febs.15292. Epub 2020 Mar 31.
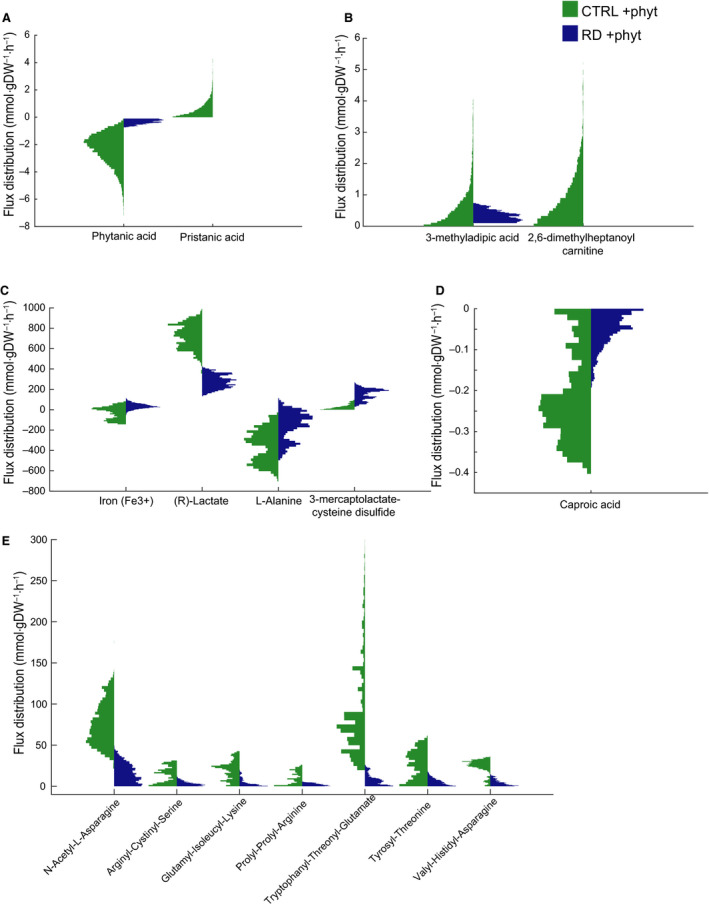
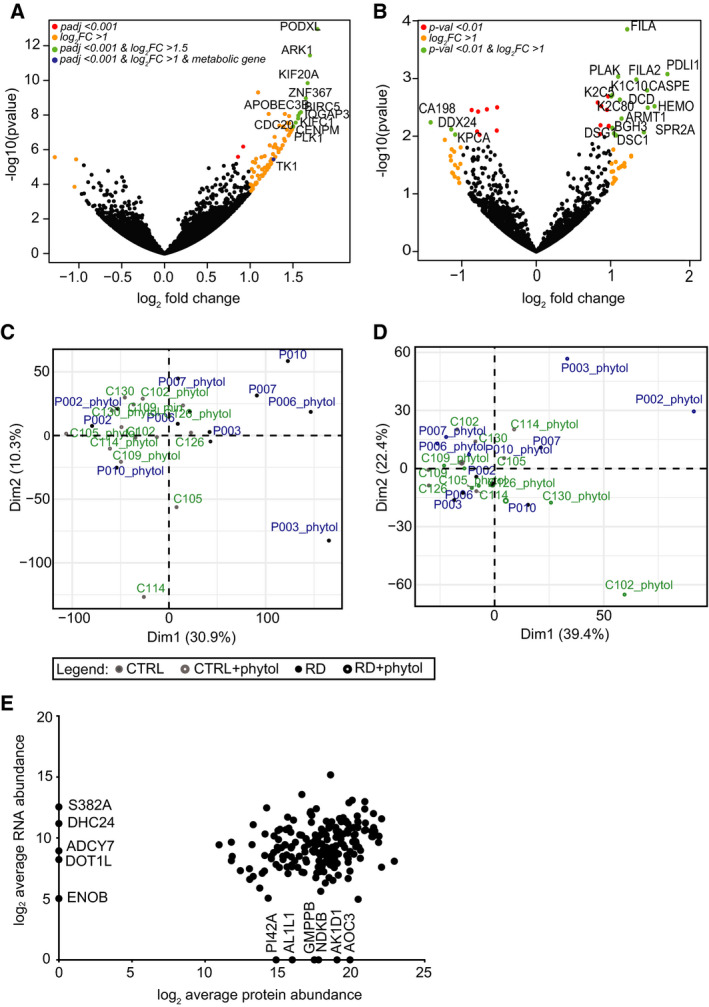
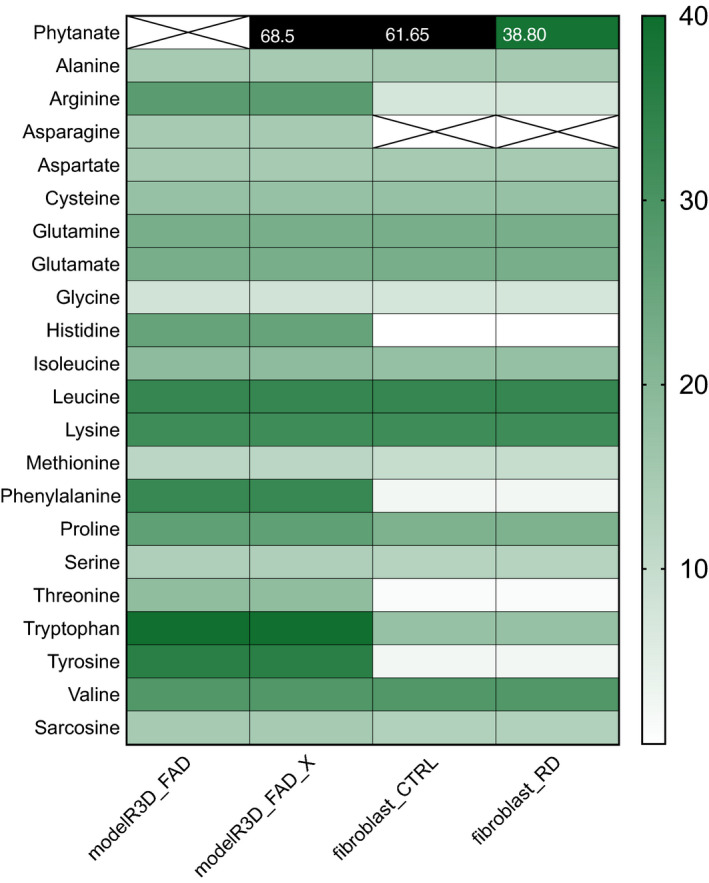
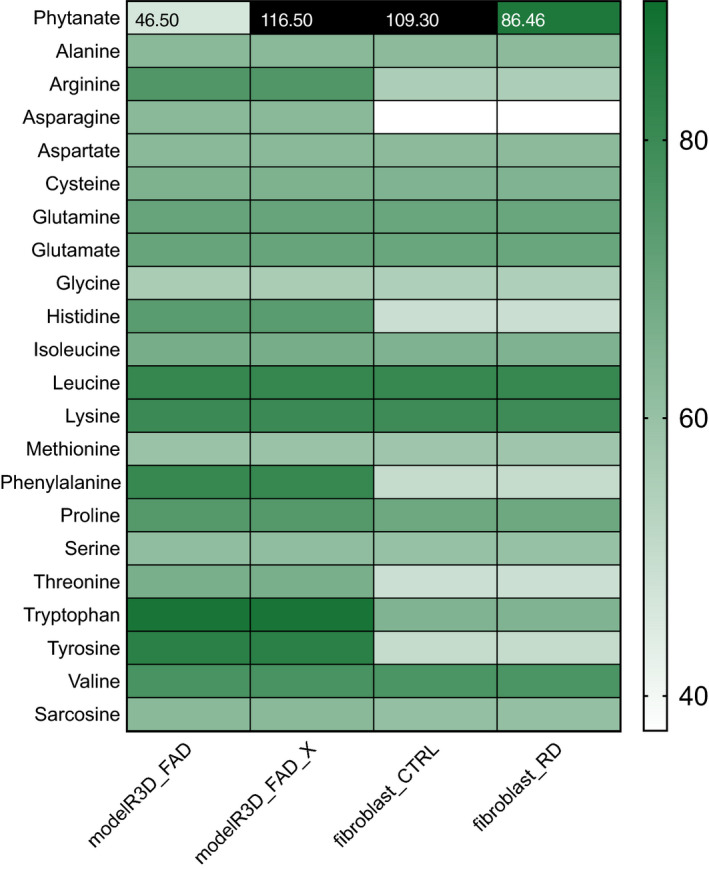
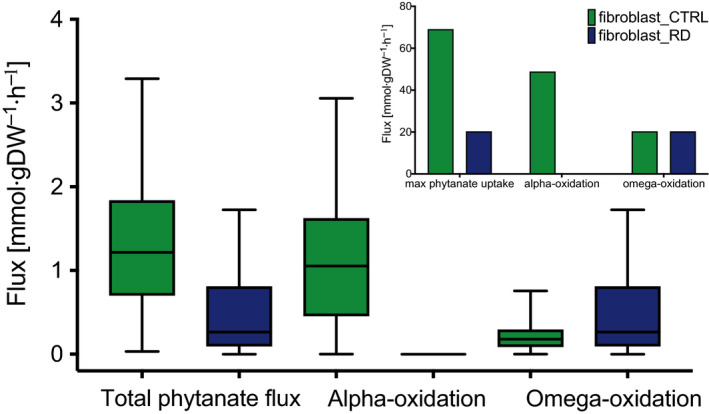
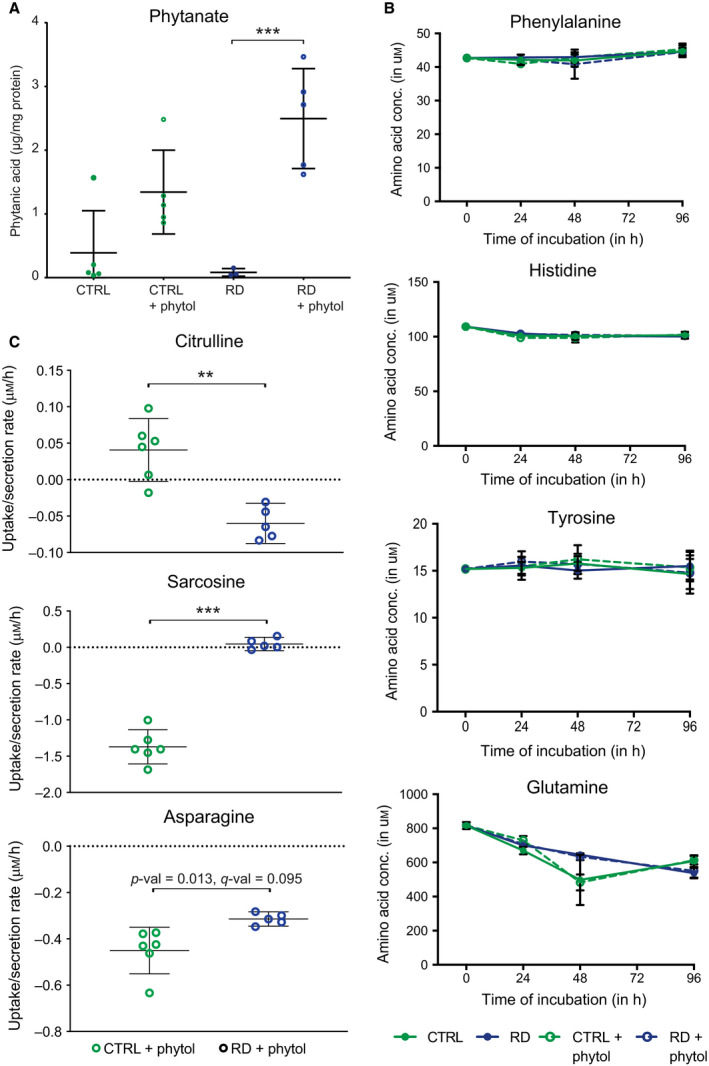
Refsum disease (RD) is an inborn error of metabolism that is characterised by a defect in peroxisomal α-oxidation of the branched-chain fatty acid phytanic acid. The disorder presents with late-onset progressive retinitis pigmentosa and polyneuropathy and can be diagnosed biochemically by elevated levels of phytanate in plasma and tissues of patients. To date, no cure exists for RD, but phytanate levels in patients can be reduced by plasmapheresis and a strict diet. In this study, we reconstructed a fibroblast-specific genome-scale model based on the recently published, FAD-curated model, based on Recon3D reconstruction. We used transcriptomics (available via GEO database with identifier GSE138379), metabolomics and proteomics (available via ProteomeXchange with identifier PXD015518) data, which we obtained from healthy controls and RD patient fibroblasts incubated with phytol, a precursor of phytanic acid. Our model correctly represents the metabolism of phytanate and displays fibroblast-specific metabolic functions. Using this model, we investigated the metabolic phenotype of RD at the genome scale, and we studied the effect of phytanate on cell metabolism. We identified 53 metabolites that were predicted to discriminate between healthy and RD patients, several of which with a link to amino acid metabolism. Ultimately, these insights in metabolic changes may provide leads for pathophysiology and therapy. DATABASES: Transcriptomics data are available via GEO database with identifier GSE138379, and proteomics data are available via ProteomeXchange with identifier PXD015518.
Refsum 病(RD)是一种先天性代谢缺陷病,其特征是支链脂肪酸植烷酸的过氧化物酶体 α-氧化缺陷。该疾病表现为迟发性进行性视网膜色素变性和多发性神经病,可通过患者血浆和组织中植烷酸水平升高进行生化诊断。迄今为止,RD 尚无治愈方法,但可通过血浆置换和严格饮食降低患者的植烷酸水平。在这项研究中,我们根据最近发表的、经 FAD 编辑的模型,基于 Recon3D 重建,构建了一个基于成纤维细胞的基因组规模模型。我们使用了转录组学(可通过 GEO 数据库获得,标识符为 GSE138379)、代谢组学和蛋白质组学(可通过 ProteomeXchange 获得,标识符为 PXD015518)数据,这些数据是从用植醇(植烷酸的前体)孵育的健康对照和成纤维细胞 RD 患者中获得的。我们的模型正确地代表了植烷酸的代谢,并显示出成纤维细胞特有的代谢功能。利用该模型,我们在基因组水平上研究了 RD 的代谢表型,并研究了植烷酸对细胞代谢的影响。我们鉴定出了 53 种代谢物,这些代谢物可用于区分健康患者和 RD 患者,其中一些代谢物与氨基酸代谢有关。最终,这些对代谢变化的深入了解可能为病理生理学和治疗提供线索。数据库:转录组学数据可通过 GEO 数据库获得,标识符为 GSE138379,蛋白质组学数据可通过 ProteomeXchange 获得,标识符为 PXD015518。