Department of Medical Biophysics, Institute of Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Pomorska 141/143, 90-236, Lodz, Poland.
ŁUKASIEWICZ Research Network-Institute of Biotechnology and Antibiotics, 5 Staroscinska St., 02-516, Warsaw, Poland.
Sci Rep. 2020 Mar 11;10(1):4479. doi: 10.1038/s41598-020-61436-x.
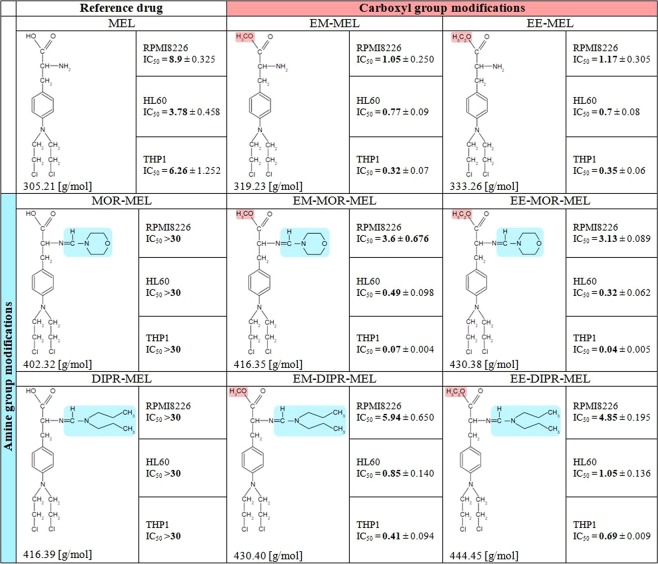
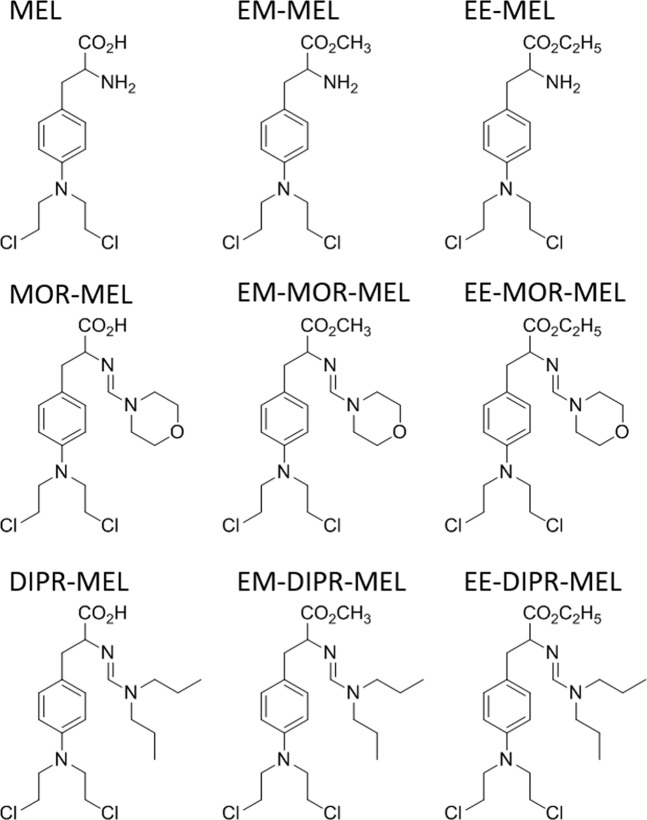
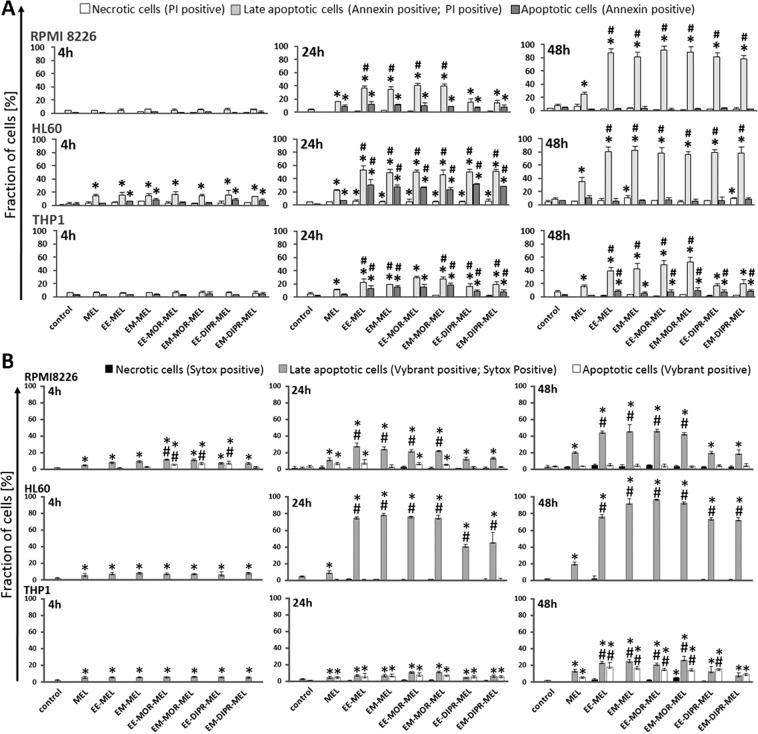
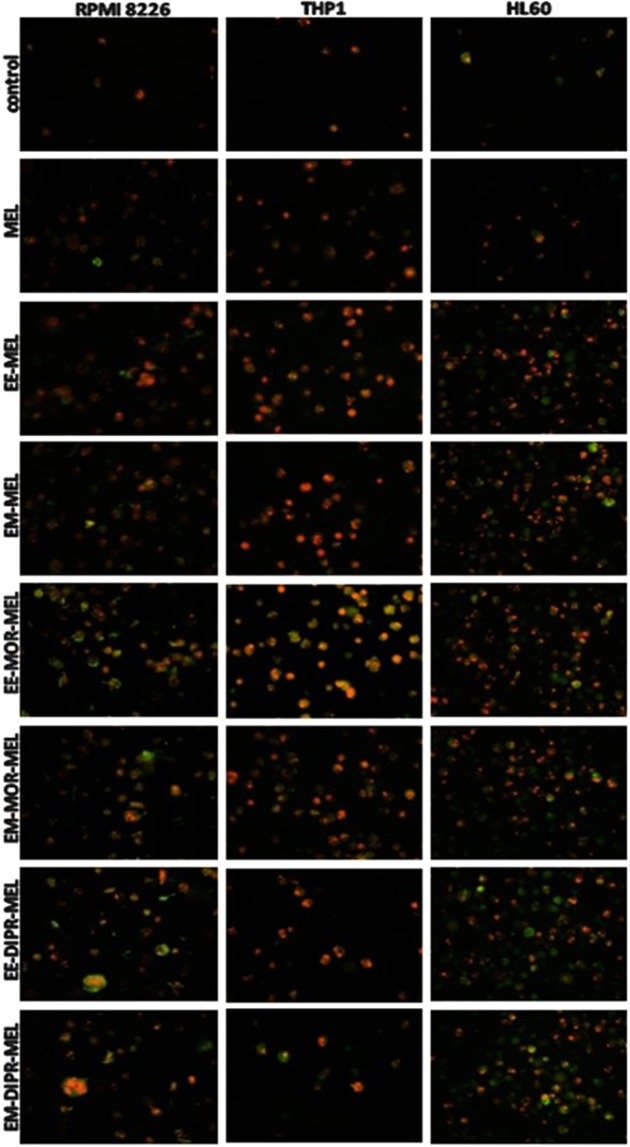
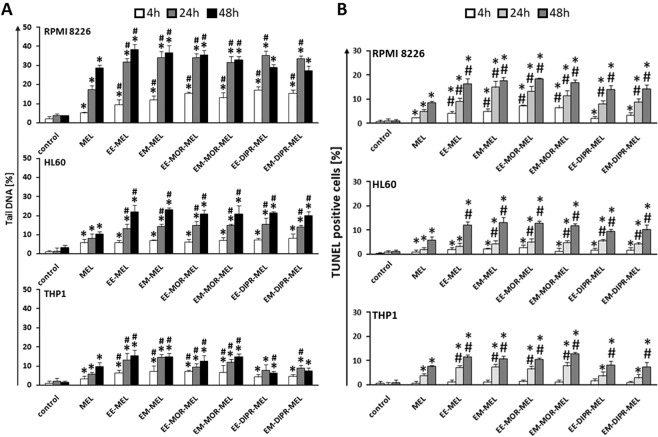
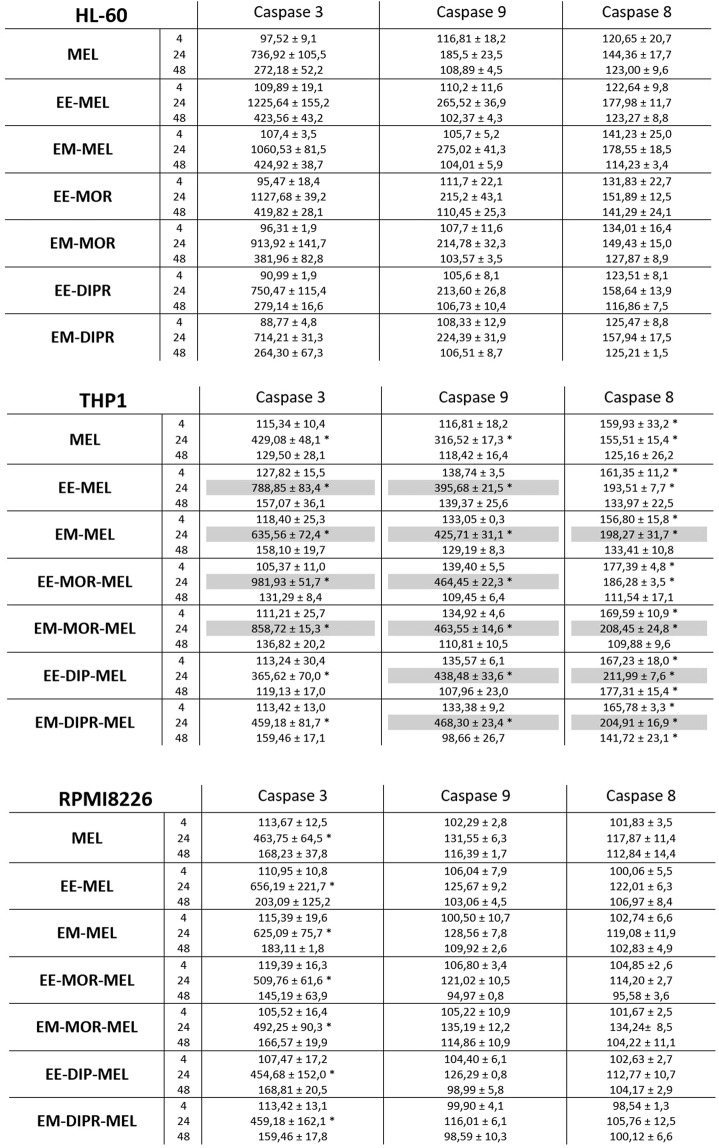
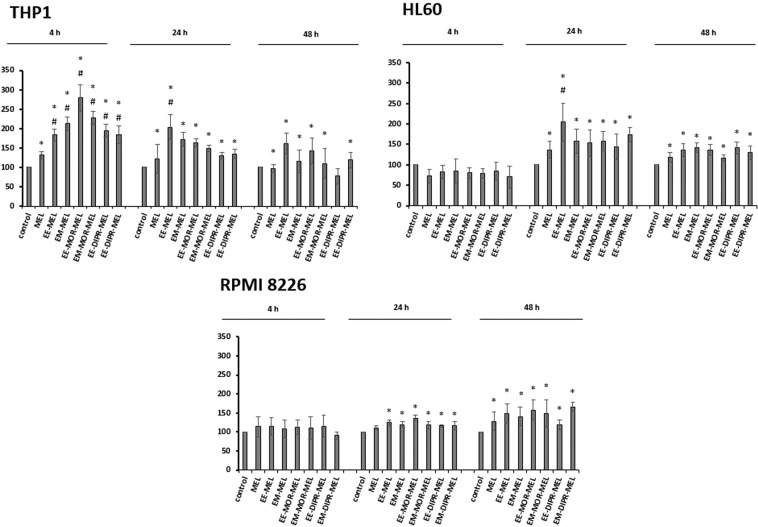
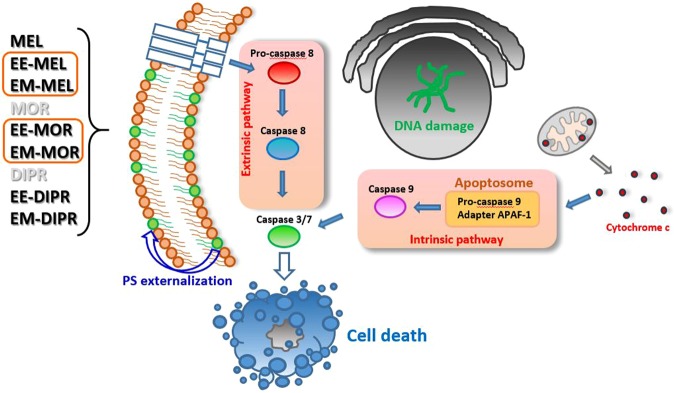
Chemical modification of known, effective drugs is one method to improve chemotherapy. Thus, the object of this study was to generate melphalan derivatives with improved cytotoxic activity in human cancer cells (RPMI8226, HL60 and THP1). Several melphalan derivatives were synthesised, modified in their two important functional groups. Nine analogues were tested, including melphalan compounds modified: only at the amino group, by replacing the amine with an amidine group containing a morpholine ring (MOR-MEL) or with an amidino group and dipropyl chain (DIPR-MEL); only at the carboxyl group to form methyl and ethyl esters of melphalan (EM-MEL, EE-MEL); and in a similar manner at both functional groups (EM-MOR-MEL, EE-MOR-MEL, EM-DIPR-MEL, EE-DIPR-MEL). Melphalan derivatives were evaluated for cytotoxicity (resazurin viability assay), genotoxicity (comet assay) and the ability to induce apoptosis (terminal deoxynucleotidyl transferase dUTP nick end labelling, TUNEL, phosphatidylserine externalisation, chromatin condensation, activity of caspases 3/7, 8 and 9 and intracellular concentration of calcium ions) in comparison with the parent drug. Almost all derivatives, with the exception of MOR-MEL and DIPR-MEL, were found to be more toxic than melphalan in all cell lines evaluated. Treatment of cultures with the derivatives generated a significant higher level of DNA breaks compared to those treated with melphalan, especially after longer incubation times. In addition, all the melphalan derivatives demonstrated a high apoptosis-inducing ability in acute monocytic and promyelocytic leukemia cells. This study showed that the mechanism of action of the tested compounds differed depending on the cell line, and allowed the selection of the most active compounds for further, more detailed investigations.
化学修饰已知的、有效的药物是改善化疗的一种方法。因此,本研究的目的是生成具有改善的细胞毒性活性的美法仑衍生物在人类癌细胞(RPMI8226、HL60 和 THP1)中。合成了几种美法仑衍生物,对其两个重要的功能基团进行了修饰。测试了 9 种类似物,包括美法仑化合物的修饰:仅在氨基上,用含有吗啉环的脒基(MOR-MEL)或脒基和二丙基链(DIPR-MEL)代替胺;仅在羧基上形成美法仑的甲酯和乙酯(EM-MEL、EE-MEL);以及以类似的方式在两个功能基团上(EM-MOR-MEL、EE-MOR-MEL、EM-DIPR-MEL、EE-DIPR-MEL)。与母体药物相比,评估了美法仑衍生物的细胞毒性(resazurin 活力测定)、遗传毒性(彗星试验)和诱导细胞凋亡的能力(末端脱氧核苷酸转移酶 dUTP 缺口末端标记,TUNEL,磷脂酰丝氨酸外化,染色质浓缩,半胱天冬酶 3/7、8 和 9 的活性和细胞内钙离子浓度)。除了 MOR-MEL 和 DIPR-MEL 之外,几乎所有的衍生物在所有评估的细胞系中都比美法仑更具毒性。与用美法仑处理的培养物相比,用这些衍生物处理的培养物产生了明显更高水平的 DNA 断裂,尤其是在更长的孵育时间后。此外,所有美法仑衍生物在急性单核细胞白血病和早幼粒细胞白血病细胞中均显示出很高的诱导细胞凋亡能力。这项研究表明,测试化合物的作用机制因细胞系而异,并允许选择最活跃的化合物进行进一步、更详细的研究。