Grinberg Nastasiya F, Orhobor Oghenejokpeme I, King Ross D
1School of Computer Science, University of Manchester, Oxford Road, Manchester, M13 9PL UK.
2Present Address: Department of Medicine, Cambridge Institute of Therapeutic Immunology & Infectious Disease, Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus, University of Cambridge, Cambridge, CB2 0AW UK.
Mach Learn. 2020;109(2):251-277. doi: 10.1007/s10994-019-05848-5. Epub 2019 Oct 23.
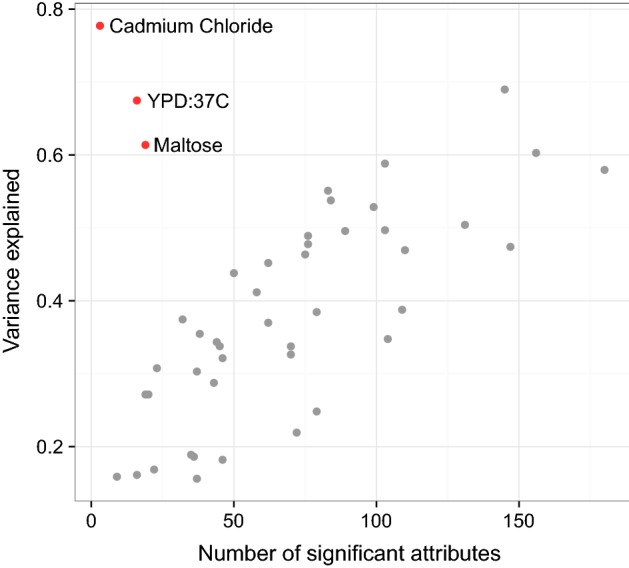
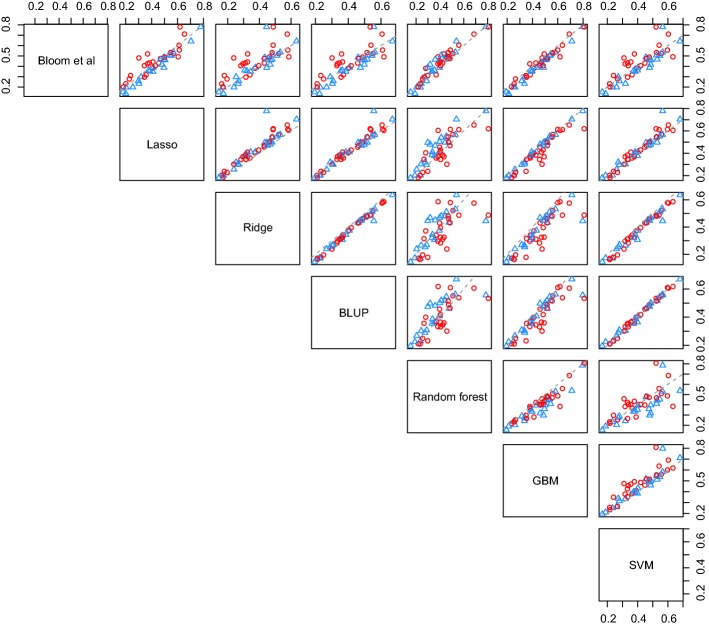
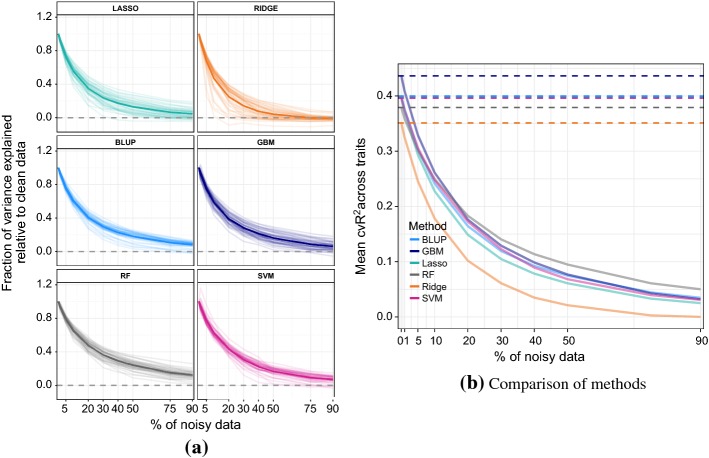
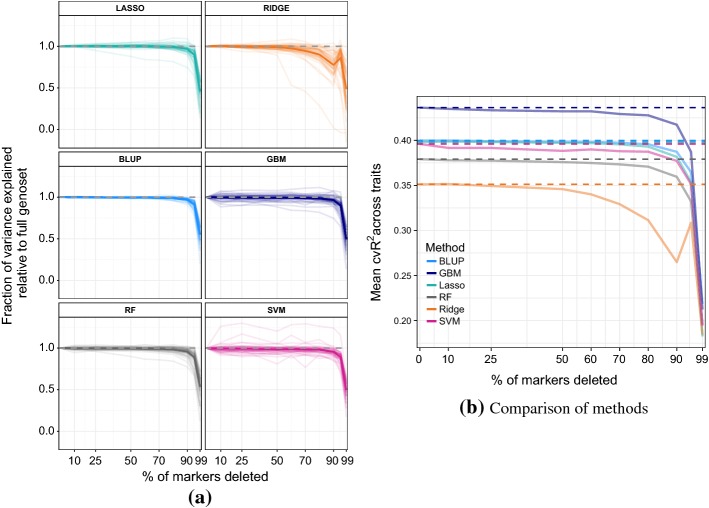
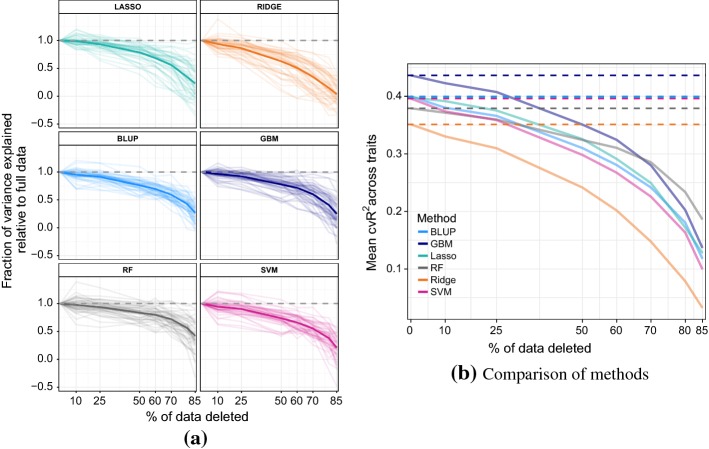
In phenotype prediction the physical characteristics of an organism are predicted from knowledge of its genotype and environment. Such studies, often called genome-wide association studies, are of the highest societal importance, as they are of central importance to medicine, crop-breeding, etc. We investigated three phenotype prediction problems: one simple and clean (yeast), and the other two complex and real-world (rice and wheat). We compared standard machine learning methods; elastic net, ridge regression, lasso regression, random forest, gradient boosting machines (GBM), and support vector machines (SVM), with two state-of-the-art classical statistical genetics methods; genomic BLUP and a two-step sequential method based on linear regression. Additionally, using the clean yeast data, we investigated how performance varied with the complexity of the biological mechanism, the amount of observational noise, the number of examples, the amount of missing data, and the use of different data representations. We found that for almost all the phenotypes considered, standard machine learning methods outperformed the methods from classical statistical genetics. On the yeast problem, the most successful method was GBM, followed by lasso regression, and the two statistical genetics methods; with greater mechanistic complexity GBM was best, while in simpler cases lasso was superior. In the wheat and rice studies the best two methods were SVM and BLUP. The most robust method in the presence of noise, missing data, etc. was random forests. The classical statistical genetics method of genomic BLUP was found to perform well on problems where there was population structure. This suggests that standard machine learning methods need to be refined to include population structure information when this is present. We conclude that the application of machine learning methods to phenotype prediction problems holds great promise, but that determining which methods is likely to perform well on any given problem is elusive and non-trivial.
在表型预测中,生物体的物理特征是根据其基因型和环境知识来预测的。这类研究,通常称为全基因组关联研究,具有极高的社会重要性,因为它们对医学、作物育种等至关重要。我们研究了三个表型预测问题:一个简单且纯粹(酵母),另外两个复杂且贴近现实(水稻和小麦)。我们将标准机器学习方法;弹性网络、岭回归、套索回归、随机森林、梯度提升机(GBM)和支持向量机(SVM),与两种最先进的经典统计遗传学方法;基因组最佳线性无偏预测(GBLUP)和基于线性回归的两步序贯方法进行了比较。此外,利用纯净的酵母数据,我们研究了性能如何随生物机制的复杂性、观测噪声量、示例数量、缺失数据量以及不同数据表示的使用而变化。我们发现,对于几乎所有考虑的表型,标准机器学习方法都优于经典统计遗传学方法。在酵母问题上,最成功的方法是GBM,其次是套索回归,以及两种统计遗传学方法;随着机制复杂性增加,GBM最佳,而在较简单的情况下套索回归更优。在小麦和水稻研究中,最佳的两种方法是SVM和GBLUP。在存在噪声、缺失数据等情况下最稳健的方法是随机森林。发现基因组GBLUP的经典统计遗传学方法在存在群体结构的问题上表现良好。这表明,当存在群体结构信息时,标准机器学习方法需要进行改进以纳入该信息。我们得出结论,将机器学习方法应用于表型预测问题前景广阔,但确定哪种方法在任何给定问题上可能表现良好是难以捉摸且并非易事的。