LABGeM, Génomique Métabolique, CEA, Genoscope, Institut François Jacob, Université d'Évry, Université Paris-Saclay, CNRS, Evry, France.
Microbial Evolutionary Genomics, Institut Pasteur, CNRS, UMR3525, Paris, France.
PLoS Comput Biol. 2020 Mar 19;16(3):e1007732. doi: 10.1371/journal.pcbi.1007732. eCollection 2020 Mar.
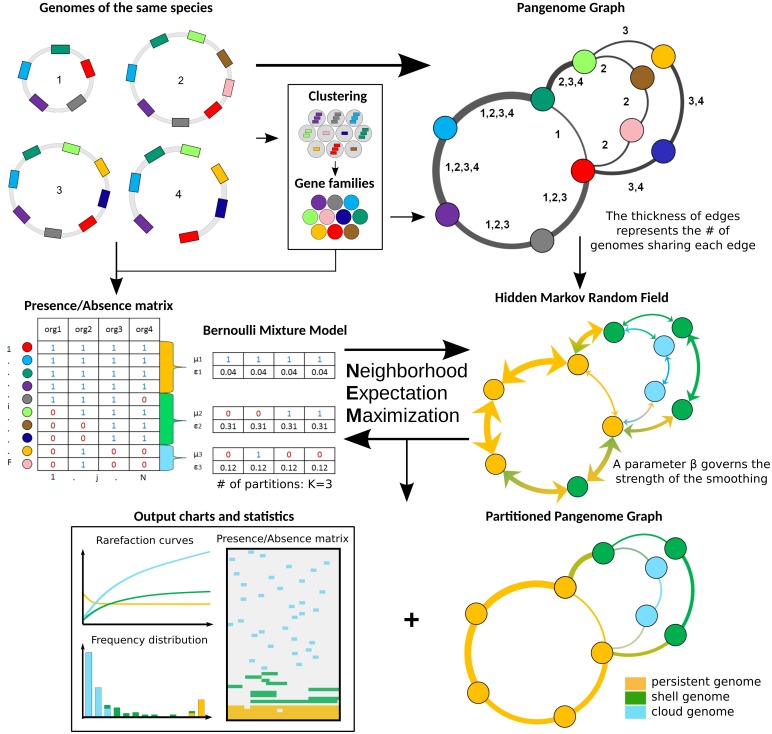
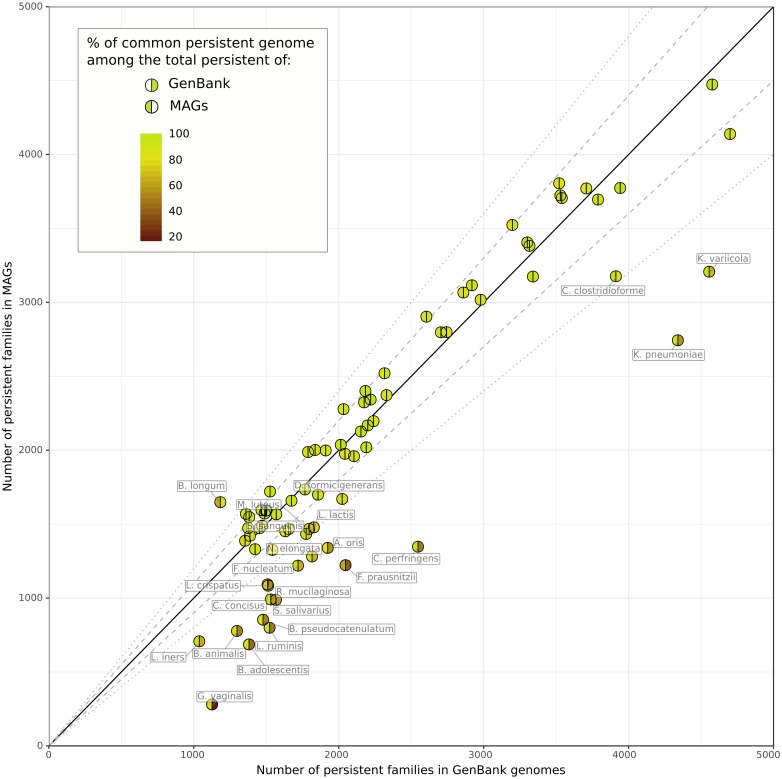
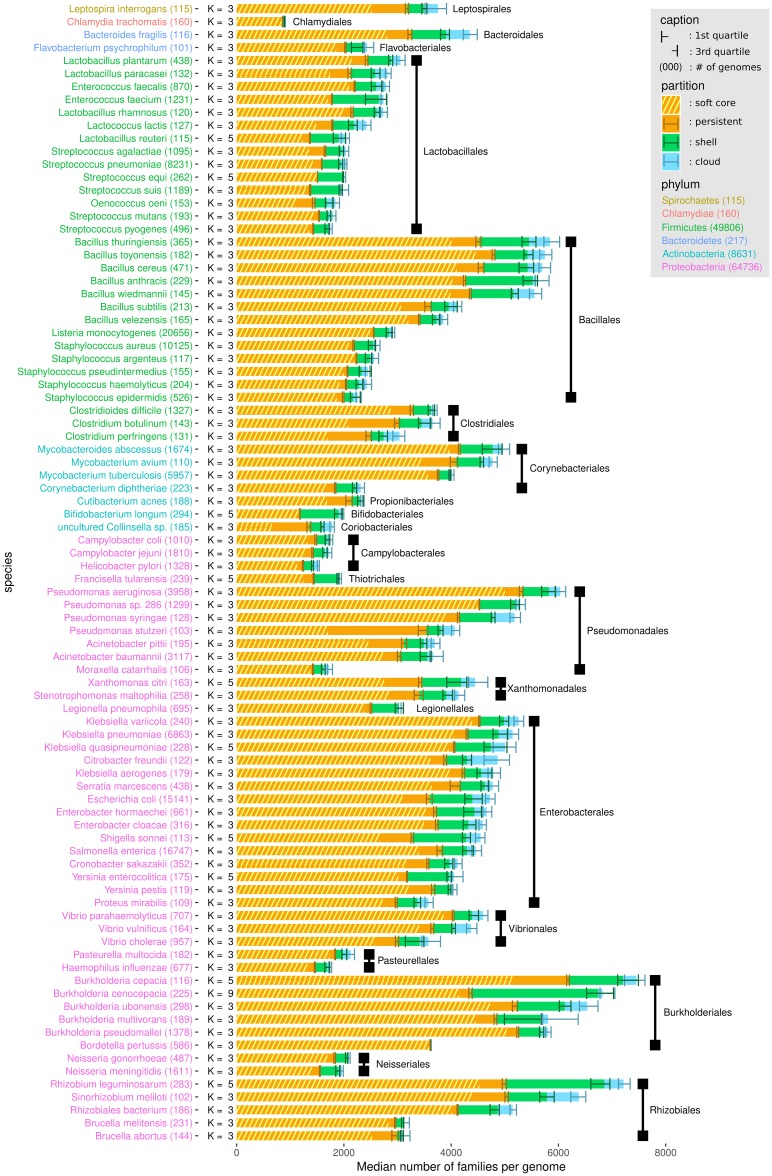
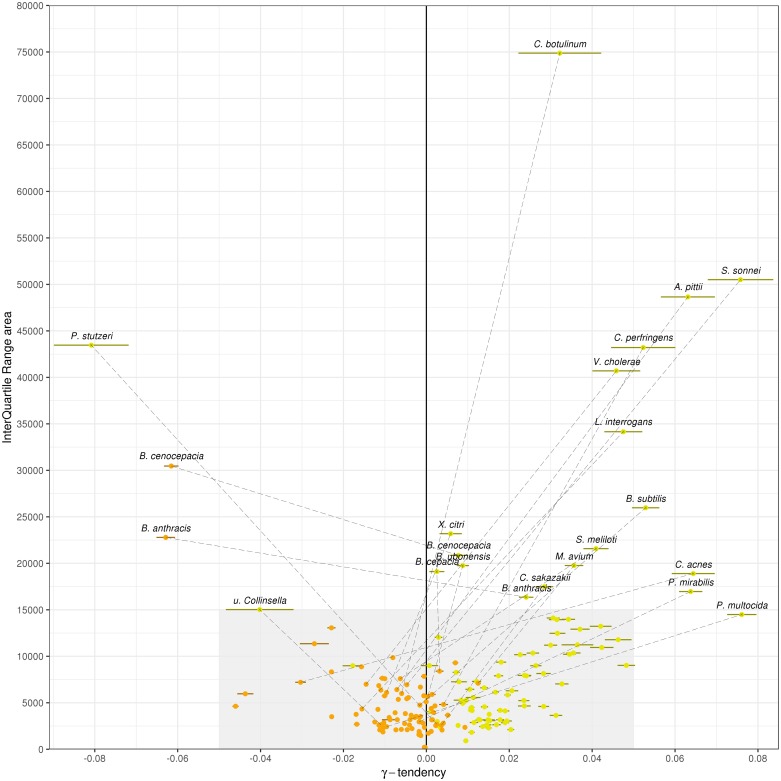
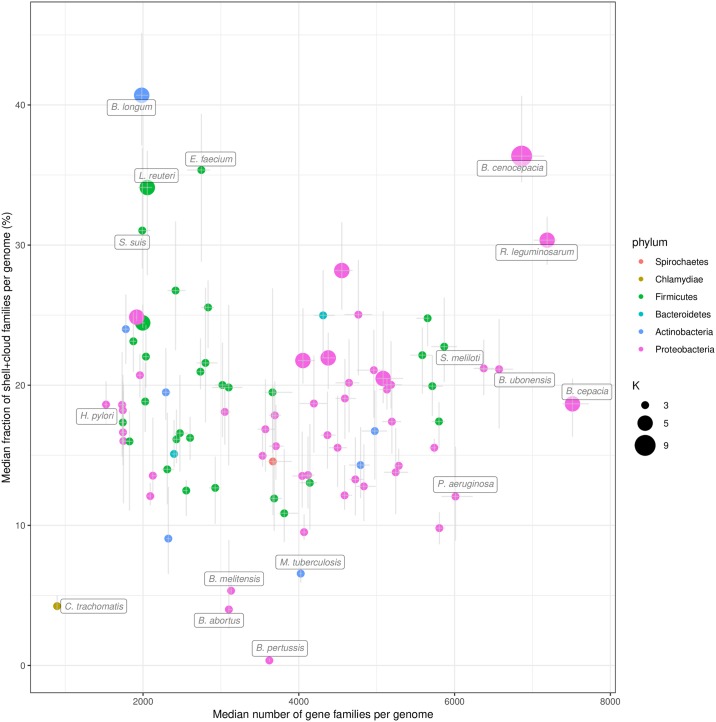
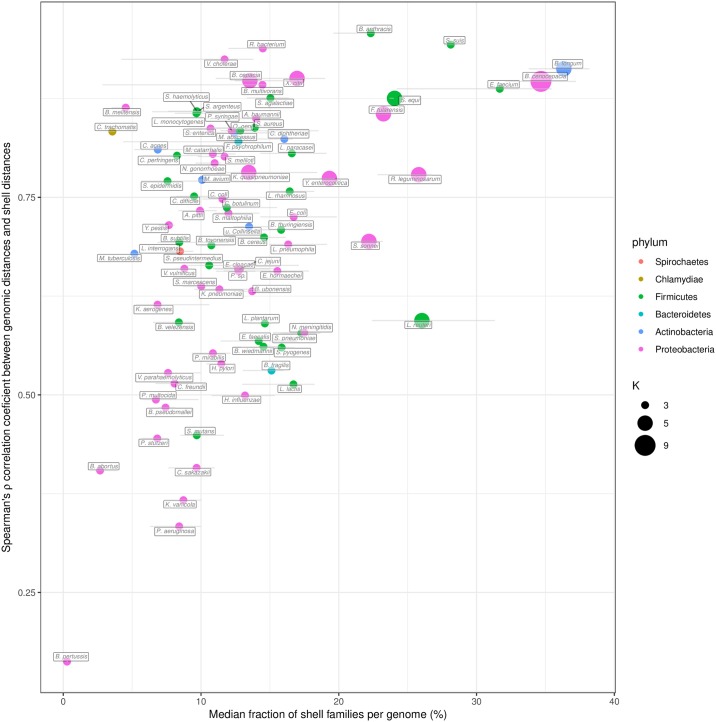
The use of comparative genomics for functional, evolutionary, and epidemiological studies requires methods to classify gene families in terms of occurrence in a given species. These methods usually lack multivariate statistical models to infer the partitions and the optimal number of classes and don't account for genome organization. We introduce a graph structure to model pangenomes in which nodes represent gene families and edges represent genomic neighborhood. Our method, named PPanGGOLiN, partitions nodes using an Expectation-Maximization algorithm based on multivariate Bernoulli Mixture Model coupled with a Markov Random Field. This approach takes into account the topology of the graph and the presence/absence of genes in pangenomes to classify gene families into persistent, cloud, and one or several shell partitions. By analyzing the partitioned pangenome graphs of isolate genomes from 439 species and metagenome-assembled genomes from 78 species, we demonstrate that our method is effective in estimating the persistent genome. Interestingly, it shows that the shell genome is a key element to understand genome dynamics, presumably because it reflects how genes present at intermediate frequencies drive adaptation of species, and its proportion in genomes is independent of genome size. The graph-based approach proposed by PPanGGOLiN is useful to depict the overall genomic diversity of thousands of strains in a compact structure and provides an effective basis for very large scale comparative genomics. The software is freely available at https://github.com/labgem/PPanGGOLiN.
比较基因组学在功能、进化和流行病学研究中的应用需要方法来根据特定物种的存在情况对基因家族进行分类。这些方法通常缺乏多元统计模型来推断分区和最佳类别数量,并且不考虑基因组组织。我们引入了一种图结构来对泛基因组进行建模,其中节点代表基因家族,边代表基因组邻居。我们的方法名为 PPanGGOLiN,使用基于多元 Bernoulli 混合模型的期望最大化算法对节点进行分区,该模型与马尔可夫随机场相结合。这种方法考虑了图的拓扑结构和泛基因组中基因的存在/缺失情况,将基因家族分类为持久、云、一个或多个壳分区。通过分析来自 439 个物种的分离株基因组的分区泛基因组图和来自 78 个物种的宏基因组组装基因组的分区泛基因组图,我们证明了我们的方法有效地估计了持久基因组。有趣的是,它表明壳基因组是理解基因组动态的关键因素,可能是因为它反映了中间频率存在的基因如何驱动物种的适应,并且它在基因组中的比例与基因组大小无关。PPanGGOLiN 提出的基于图的方法有助于用紧凑的结构描绘数千个菌株的整体基因组多样性,并为非常大规模的比较基因组学提供有效的基础。该软件可在 https://github.com/labgem/PPanGGOLiN 上免费获得。