Lyda Hill Department of Bioinformatics, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Department of Computer Science, Stanford University, Stanford, CA, USA.
Nat Biotechnol. 2019 Aug;37(8):907-915. doi: 10.1038/s41587-019-0201-4. Epub 2019 Aug 2.
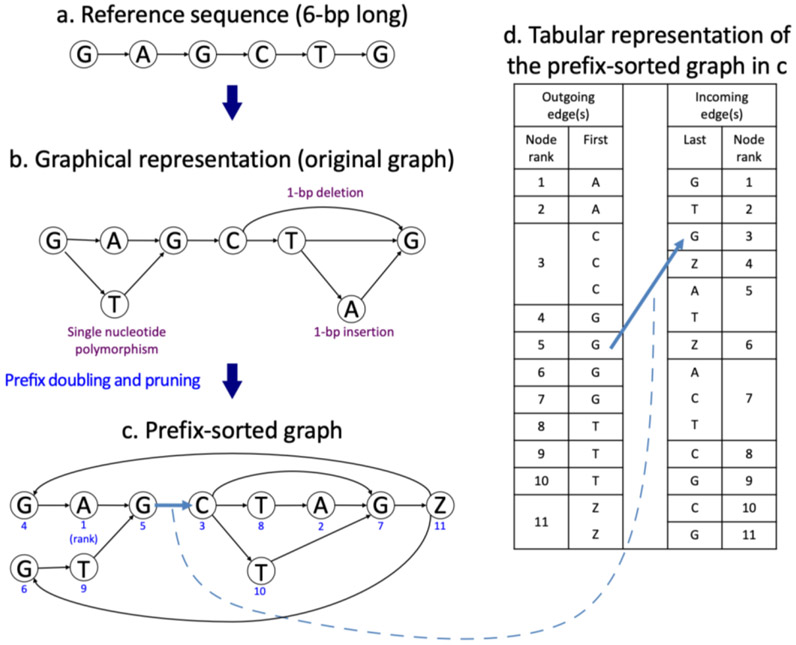
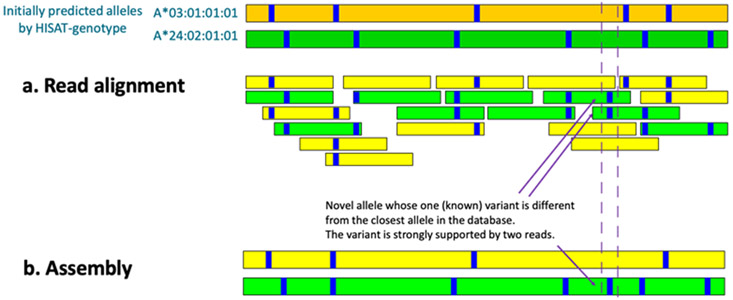
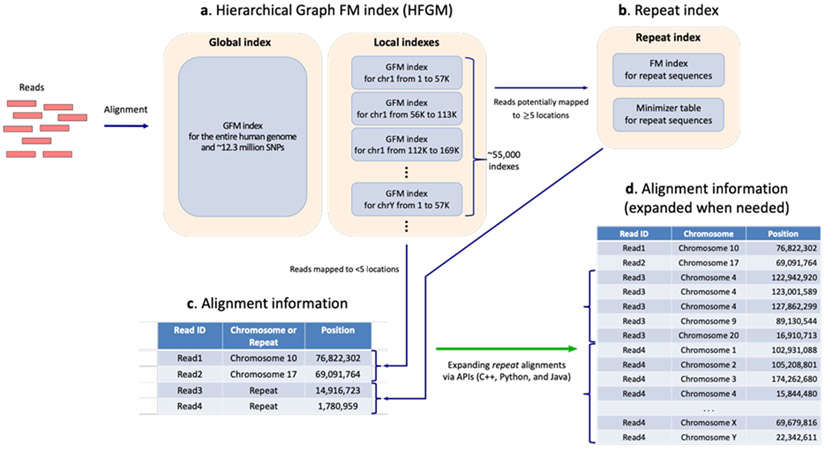
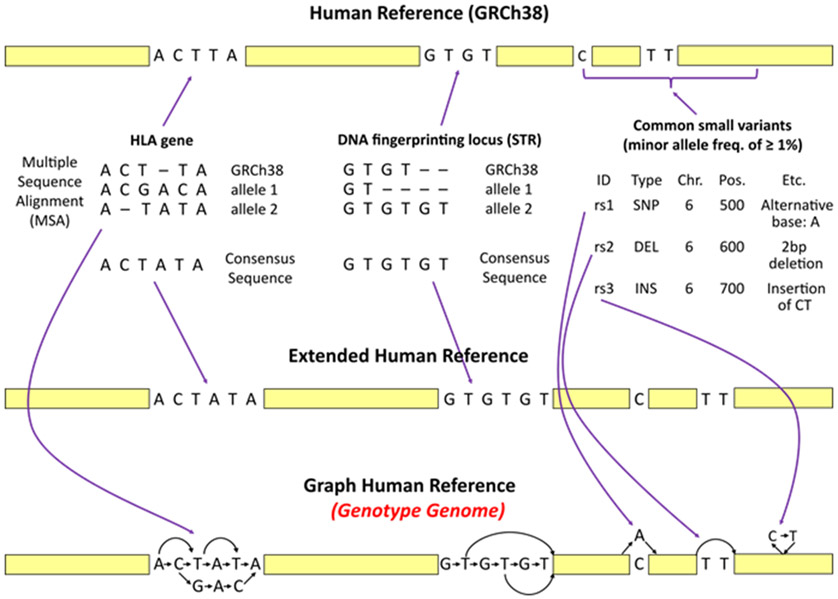
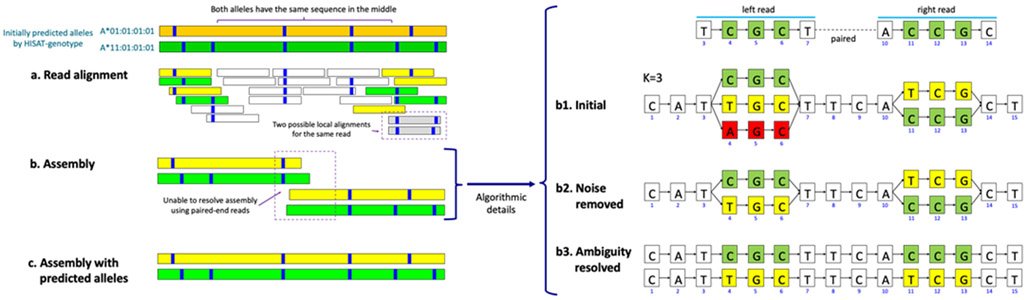
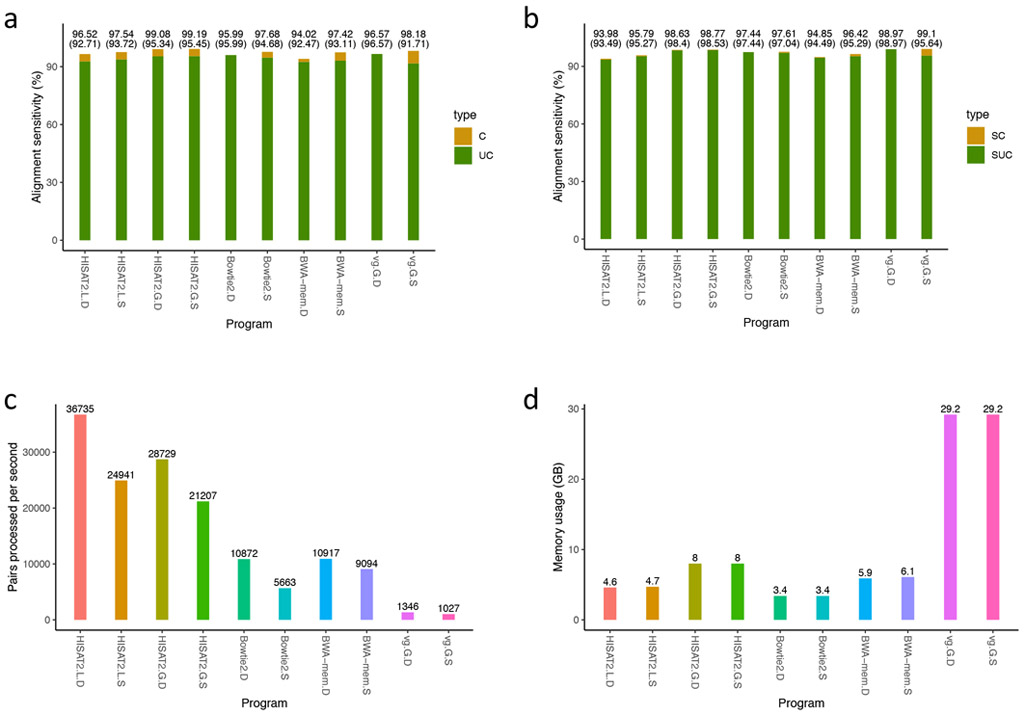
The human reference genome represents only a small number of individuals, which limits its usefulness for genotyping. We present a method named HISAT2 (hierarchical indexing for spliced alignment of transcripts 2) that can align both DNA and RNA sequences using a graph Ferragina Manzini index. We use HISAT2 to represent and search an expanded model of the human reference genome in which over 14.5 million genomic variants in combination with haplotypes are incorporated into the data structure used for searching and alignment. We benchmark HISAT2 using simulated and real datasets to demonstrate that our strategy of representing a population of genomes, together with a fast, memory-efficient search algorithm, provides more detailed and accurate variant analyses than other methods. We apply HISAT2 for HLA typing and DNA fingerprinting; both applications form part of the HISAT-genotype software that enables analysis of haplotype-resolved genes or genomic regions. HISAT-genotype outperforms other computational methods and matches or exceeds the performance of laboratory-based assays.
人类参考基因组仅代表少数个体,这限制了其用于基因分型的用途。我们提出了一种名为 HISAT2(用于转录本拼接对齐的分层索引 2)的方法,该方法可以使用 Ferragina Manzini 图索引同时对齐 DNA 和 RNA 序列。我们使用 HISAT2 来表示和搜索扩展的人类参考基因组模型,其中包含超过 1450 万个基因组变体,以及与单倍型结合的变体,这些变体被合并到用于搜索和对齐的数
据结构中。我们使用模拟和真实数据集对 HISAT2 进行基准测试,以证明我们代表基因组群体的策略,以及快速、内存高效的搜索算法,比其他方法提供更详细和准确的变体分析。我们将 HISAT2 应用于 HLA 分型和 DNA 指纹分析;这两个应用程序都是 HISAT-genotype 软件的一部分,该软件能够分析单倍型解析基因或基因组区域。HISAT-genotype 优于其他计算方法,并且与基于实验室的检测方法的性能相匹配或超过。