Sova Matej, Frlan Rok, Gobec Stanislav, Časar Zdenko
University of Ljubljana, Faculty of Pharmacy, Aškerčeva 7, Ljubljana 1000, Slovenia.
Lek Pharmaceuticals, d.d., Sandoz Development Center Slovenia, Verovškova ulica 57, Ljubljana SI-1526, Slovenia.
ACS Omega. 2020 Mar 3;5(10):5356-5364. doi: 10.1021/acsomega.9b04393. eCollection 2020 Mar 17.
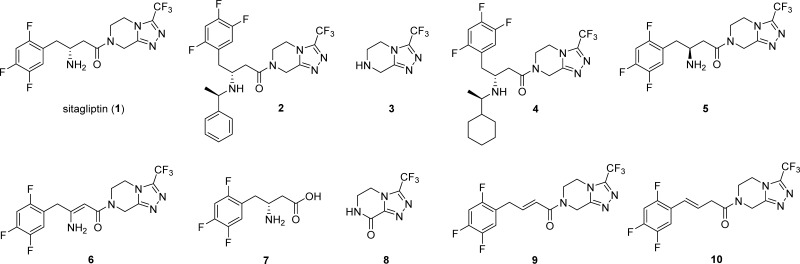
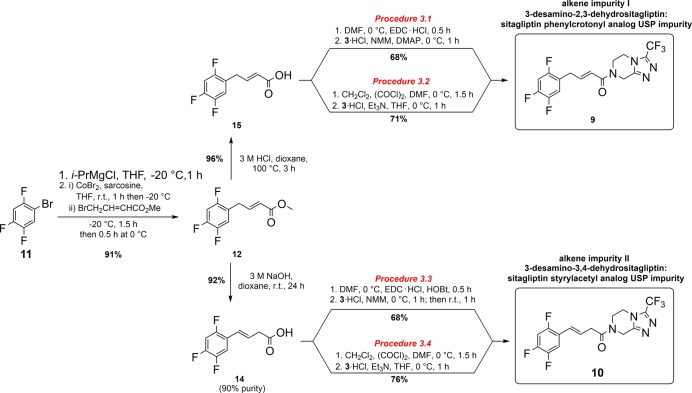
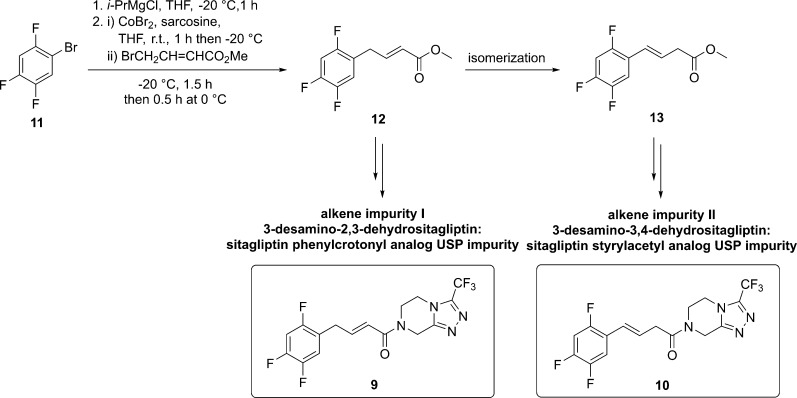
Various organic impurities (starting materials, reagents, intermediates, degradation products, by-products, and side products) could be present in active pharmaceutical ingredients affecting their qualities, safeties, and efficacies. Herein, we present the efficient syntheses of two United States Pharmacopeia impurities of an antidiabetic drug sitagliptin, a potent and orally active dipeptidyl peptidase IV inhibitor: 3-desamino-2,3-dehydrositagliptin and 3-desamino-3,4-dehydrositagliptin. Our three-step synthetic approach is based on the efficient cobalt-catalyzed cross-coupling reaction of 1-bromo-2,4,5-trifluorobenzene and methyl 4-bromocrotonate in the first step, followed by hydrolysis of corresponding ester with 3 M HCl to ()-(2,4,5-trifluorophenyl)but-2-enoic acid in high overall yield, whereas the reaction with 3 M NaOH resulted in the carbon-carbon double bond regio-isomerization and hydrolysis to give the ()-(2,4,5-trifluorophenyl)but-3-enoic acid in 92% yield. Both acid derivatives were converted to title compounds via the amide bond formation with 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-]pyrazine. Extensive screening of coupling/activation reagents, bases, and solvents reviled that the amide bond is formed the most efficiently using the (COCl)/EtN in THF or alternatively EDC/NMM/(DMAP or HOBt) in DMF obtaining the title compounds in 68-76% yields and providing the overall yields for the three-step process in the range of 57-64% on a gram scale. The presented study also demonstrates the importance of a proper selection of solvent, base, and coupling/activating reagent for amide bond formation using Michael acceptor-type allylbenzene derivatives as coupling partners to minimize the carbon-carbon double bond regio-isomerization.
各种有机杂质(起始原料、试剂、中间体、降解产物、副产物和杂质)可能存在于活性药物成分中,影响其质量、安全性和有效性。在此,我们展示了两种美国药典中抗糖尿病药物西他列汀(一种强效口服活性二肽基肽酶IV抑制剂)杂质的高效合成方法:3-去氨基-2,3-脱氢西他列汀和3-去氨基-3,4-脱氢西他列汀。我们的三步合成方法第一步基于1-溴-2,4,5-三氟苯与4-溴巴豆酸甲酯的高效钴催化交叉偶联反应,随后用3 M盐酸水解相应的酯,以较高的总收率得到()-(2,4,5-三氟苯基)丁-2-烯酸,而与3 M氢氧化钠反应则导致碳-碳双键区域异构化并水解,以92%的收率得到()-(2,4,5-三氟苯基)丁-3-烯酸。两种酸衍生物通过与3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-]吡嗪形成酰胺键转化为目标化合物。对偶联/活化试剂、碱和溶剂进行广泛筛选发现,使用(COCl)/EtN在四氢呋喃中或EDC/NMM/(DMAP或HOBt)在N,N-二甲基甲酰胺中最有效地形成酰胺键,以68-76%的收率得到目标化合物,并在克级规模上提供三步过程的总收率在57-64%范围内。本研究还证明了使用迈克尔受体型烯丙基苯衍生物作为偶联伙伴形成酰胺键时,正确选择溶剂、碱和偶联/活化试剂对于最小化碳-碳双键区域异构化的重要性。