Metabolic Genomics, Genoscope, Institut de Biologie François Jacob, CEA, CNRS, Univ Evry, Université Paris Saclay, 91000 Evry, France.
Research Federation for the Study of Global Ocean Systems Ecology and Evolution, FR2022/Tara Oceans GOSEE, 75016 Paris, France.
Genome Res. 2020 Apr;30(4):647-659. doi: 10.1101/gr.253070.119. Epub 2020 Mar 23.
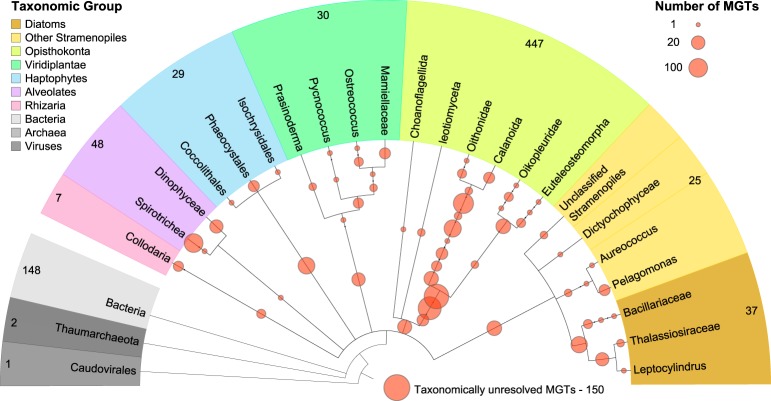
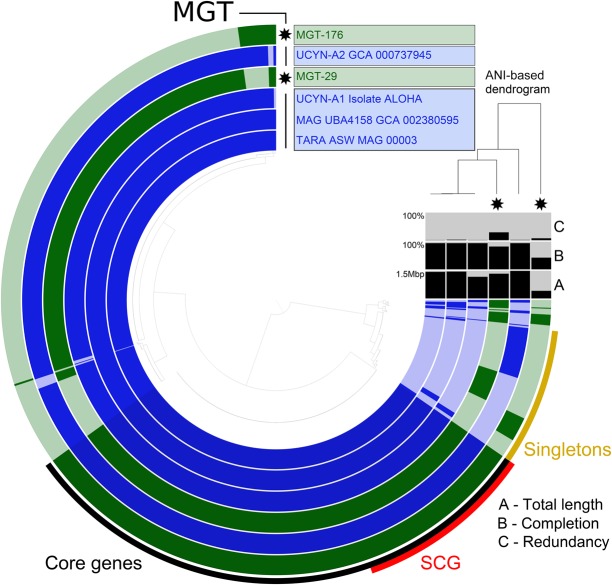
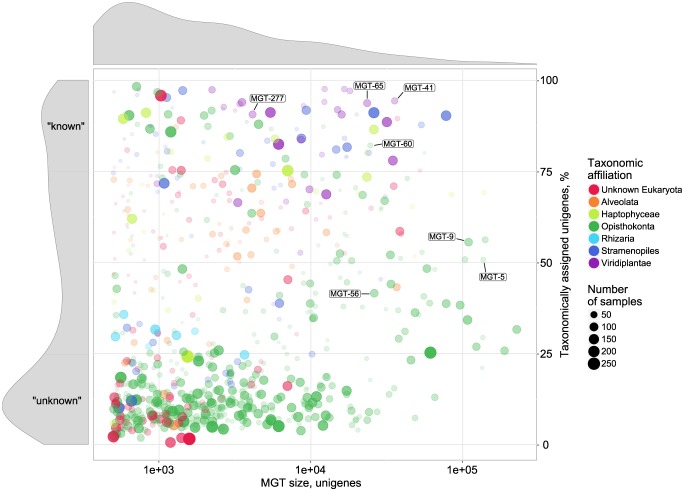
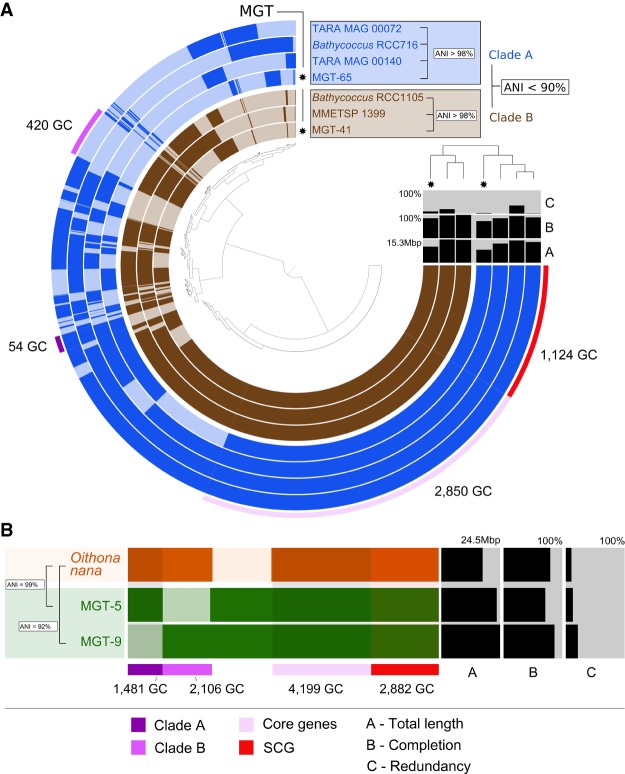
Large-scale metagenomic and metatranscriptomic data analyses are often restricted by their gene-centric approach, limiting the ability to understand organismal and community biology. De novo assembly of large and mosaic eukaryotic genomes from complex meta-omics data remains a challenging task, especially in comparison with more straightforward bacterial and archaeal systems. Here, we use a transcriptome reconstruction method based on clustering co-abundant genes across a series of metagenomic samples. We investigated the co-abundance patterns of ∼37 million eukaryotic unigenes across 365 metagenomic samples collected during the Oceans expeditions to assess the diversity and functional profiles of marine plankton. We identified ∼12,000 co-abundant gene groups (CAGs), encompassing ∼7 million unigenes, including 924 metagenomics-based transcriptomes (MGTs, CAGs larger than 500 unigenes). We demonstrated the biological validity of the MGT collection by comparing individual MGTs with available references. We identified several key eukaryotic organisms involved in dimethylsulfoniopropionate (DMSP) biosynthesis and catabolism in different oceanic provinces, thus demonstrating the potential of the MGT collection to provide functional insights on eukaryotic plankton. We established the ability of the MGT approach to capture interspecies associations through the analysis of a nitrogen-fixing haptophyte-cyanobacterial symbiotic association. This MGT collection provides a valuable resource for analyses of eukaryotic plankton in the open ocean by giving access to the genomic content and functional potential of many ecologically relevant eukaryotic species.
大规模的宏基因组学和宏转录组学数据分析通常受到其基因中心方法的限制,限制了理解生物个体和群落生物学的能力。从复杂的元组学数据中从头组装大型镶嵌真核生物基因组仍然是一项具有挑战性的任务,特别是与更简单的细菌和古菌系统相比。在这里,我们使用了一种基于跨一系列宏基因组样本聚类共丰度基因的转录组重建方法。我们调查了约 3700 万个真核生物 unigenes 在 365 个宏基因组样本中的共丰度模式,这些样本是在海洋考察期间收集的,用于评估海洋浮游生物的多样性和功能特征。我们确定了约 12000 个共丰度基因群(CAG),包含约 700 万个 unigenes,包括 924 个基于宏基因组学的转录组(CAG 大于 500 个 unigenes)。我们通过将单个 MGT 与可用的参考资料进行比较,证明了 MGT 集合的生物学有效性。我们确定了几个关键的真核生物,它们参与不同海洋区域的二甲基硫代丙酸(DMSP)生物合成和分解代谢,从而证明了 MGT 集合在真核浮游生物功能研究方面的潜力。我们通过分析固氮甲藻-蓝细菌共生体关联,证明了 MGT 方法捕捉种间关联的能力。该 MGT 集合通过访问许多生态相关的真核生物物种的基因组内容和功能潜力,为分析开阔海域中的真核浮游生物提供了有价值的资源。