State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201210, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
Acta Pharmacol Sin. 2020 Oct;41(10):1366-1376. doi: 10.1038/s41401-020-0389-3. Epub 2020 Mar 31.
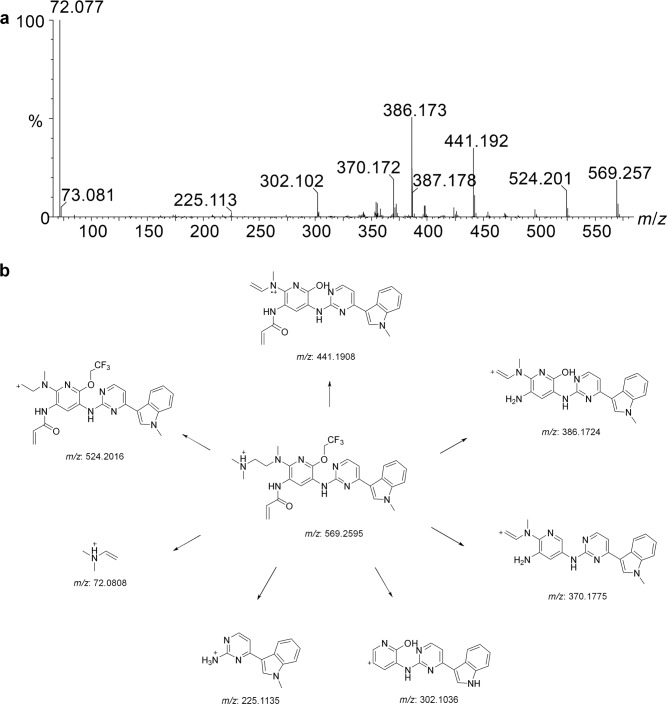
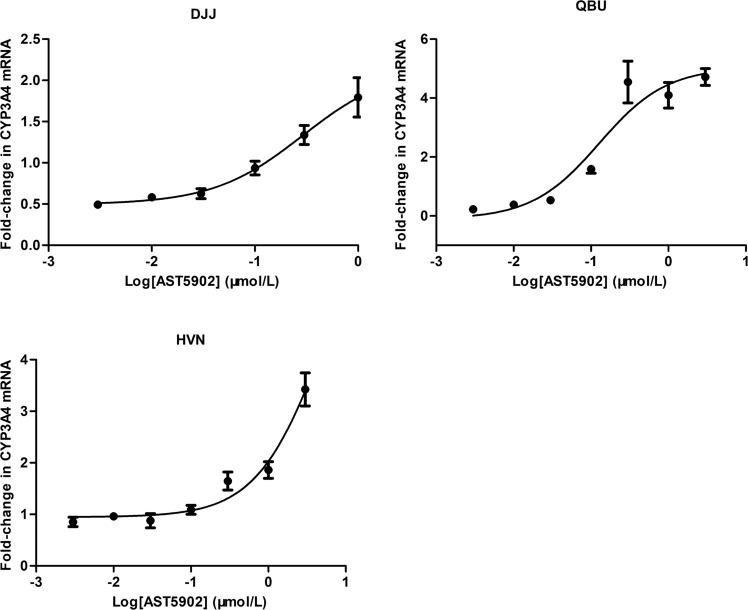
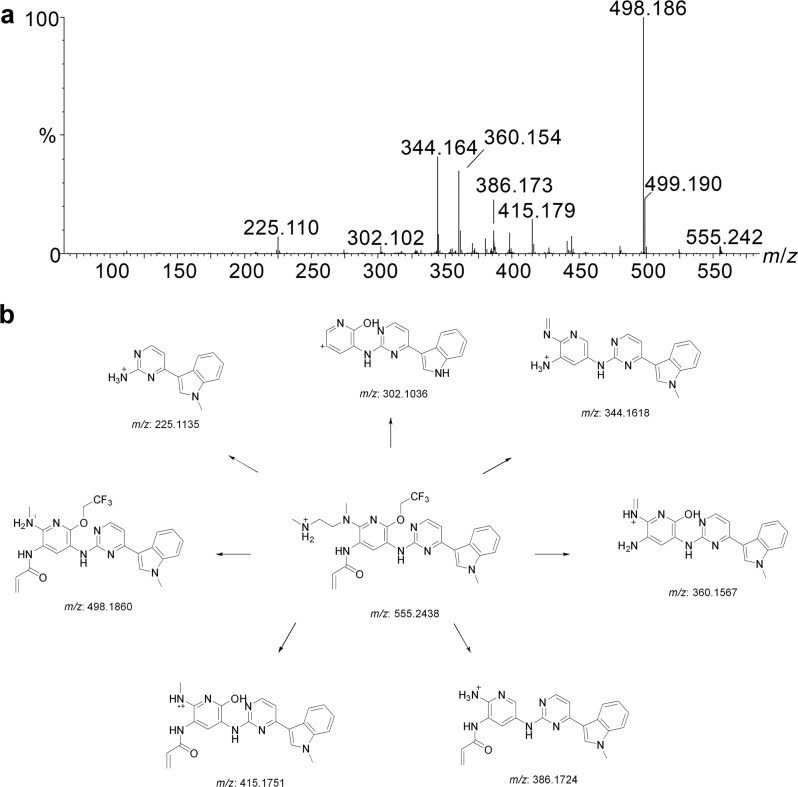
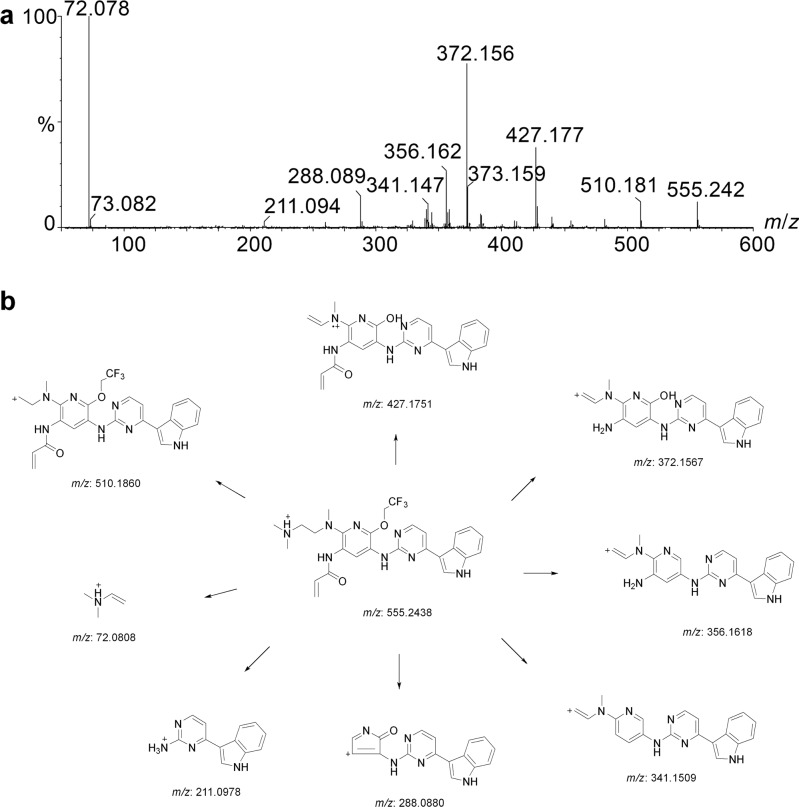
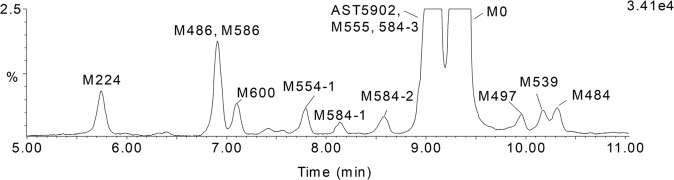
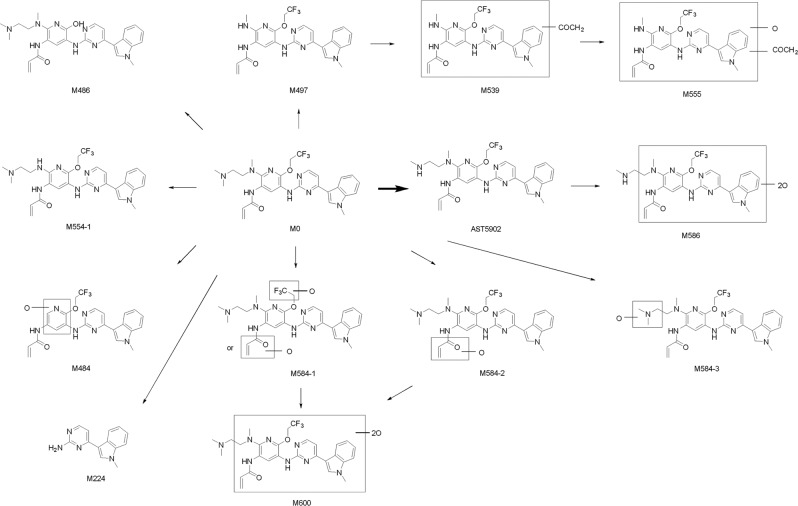
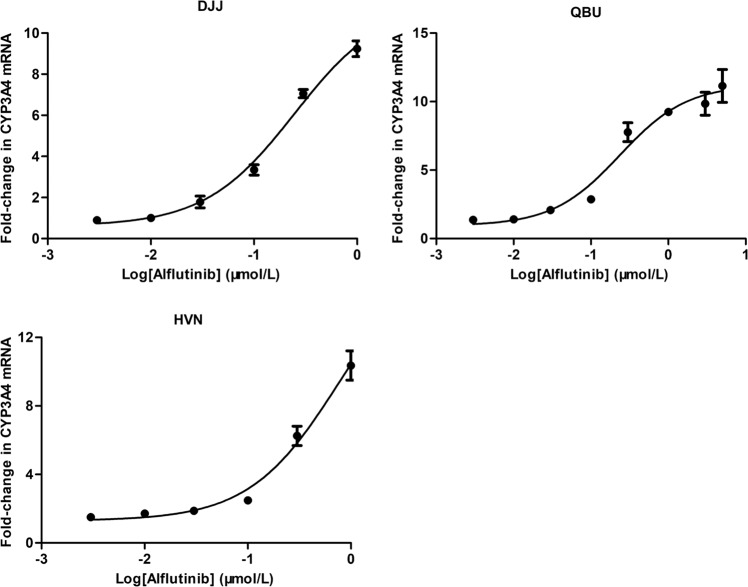
Alflutinib (AST2818) is a third-generation epidermal growth factor receptor (EGFR) inhibitor that inhibits both EGFR-sensitive mutations and T790M mutations. Previous study has shown that after multiple dosages, alflutinib exhibits nonlinear pharmacokinetics and displays a time- and dose-dependent increase in the apparent clearance, probably due to its self-induction of cytochrome P450 (CYP) enzyme. In this study, we investigated the CYP isozymes involved in the metabolism of alflutinib and evaluated the enzyme inhibition and induction potential of alflutinib and its metabolites. The data showed that alflutinib in human liver microsomes (HLMs) was metabolized mainly by CYP3A4, which could catalyze the formation of AST5902. Alflutinib did not inhibit CYP isozymes in HLMs but could induce CYP3A4 in human hepatocytes. Rifampin is a known strong CYP3A4 inducer and is recommended by the FDA as a positive control in the CYP3A4 induction assay. We found that the induction potential of alflutinib was comparable to that of rifampin. The E of CYP3A4 induction by alflutinib in three lots of human hepatocytes were 9.24-, 11.2-, and 10.4-fold, while the fold-induction of rifampin (10 μM) were 7.22-, 19.4- and 9.46-fold, respectively. The EC of alflutinib-induced CYP3A4 mRNA expression was 0.25 μM, which was similar to that of rifampin. In addition, AST5902 exhibited much weak CYP3A4 induction potential compared to alflutinib. Given the plasma exposure of alflutinib and AST5902, both are likely to affect the pharmacokinetics of CYP3A4 substrates. Considering that alflutinib is a CYP3A4 substrate and a potent CYP3A4 inducer, drug-drug interactions are expected during alflutinib treatment.
阿法替尼(AST2818)是第三代表皮生长因子受体(EGFR)抑制剂,既能抑制 EGFR 敏感突变,又能抑制 T790M 突变。之前的研究表明,阿法替尼多次给药后表现出非线性药代动力学特征,且其表观清除率呈时间和剂量依赖性增加,这可能是由于其对细胞色素 P450(CYP)酶的自身诱导。在本研究中,我们研究了参与阿法替尼代谢的 CYP 同工酶,并评估了阿法替尼及其代谢物的酶抑制和诱导潜力。研究数据显示,人肝微粒体(HLMs)中的阿法替尼主要通过 CYP3A4 代谢,能够催化 AST5902 的形成。阿法替尼对 HLMs 中的 CYP 同工酶没有抑制作用,但能在人原代肝细胞中诱导 CYP3A4。利福平是一种已知的强效 CYP3A4 诱导剂,被 FDA 推荐为 CYP3A4 诱导检测的阳性对照药物。我们发现,阿法替尼的诱导潜力与利福平相当。在三批人原代肝细胞中,阿法替尼对 CYP3A4 的诱导 E 值分别为 9.24、11.2 和 10.4 倍,而利福平(10 μM)的诱导倍数分别为 7.22、19.4 和 9.46 倍。阿法替尼诱导 CYP3A4mRNA 表达的 EC 值为 0.25 μM,与利福平相似。此外,AST5902 对 CYP3A4 的诱导潜力明显弱于阿法替尼。鉴于阿法替尼和 AST5902 的血浆暴露量,两者均可能影响 CYP3A4 底物的药代动力学。考虑到阿法替尼是 CYP3A4 的底物和强效 CYP3A4 诱导剂,在阿法替尼治疗期间预计会发生药物相互作用。