Institute of Pharmacology & Toxicology, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, Zhejiang, China.
Department of Oncology, Key Laboratory of Clinical Cancer Pharmacology and Toxicology Research of Zhejiang Province, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, Hangzhou, 310006, Zhejiang, China.
Cell Res. 2020 Sep;30(9):779-793. doi: 10.1038/s41422-020-0309-6. Epub 2020 Apr 15.
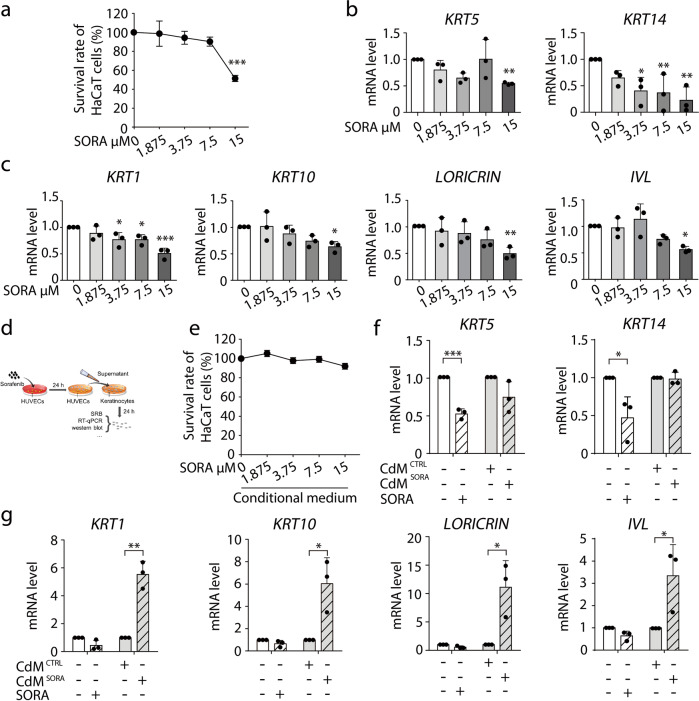
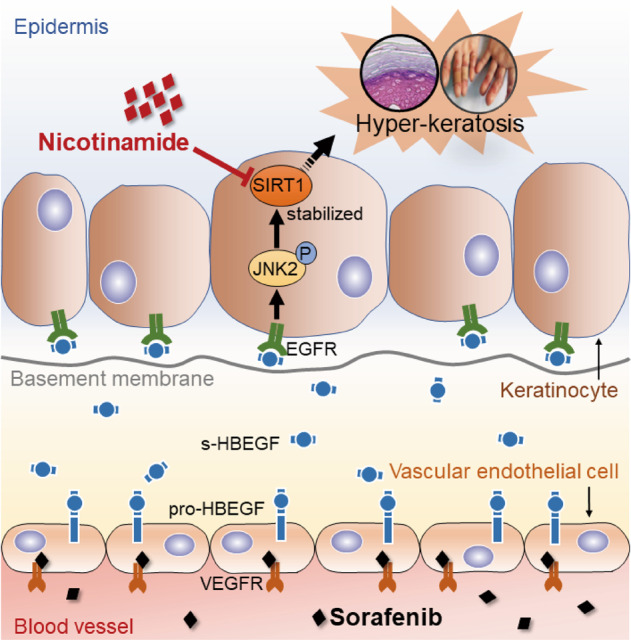
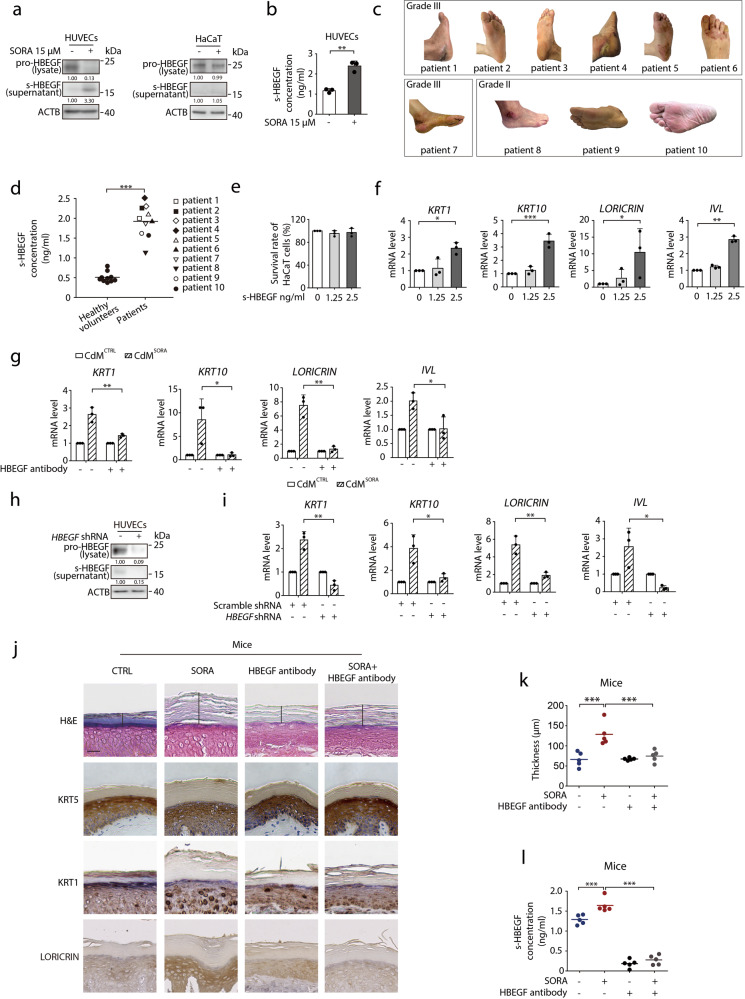
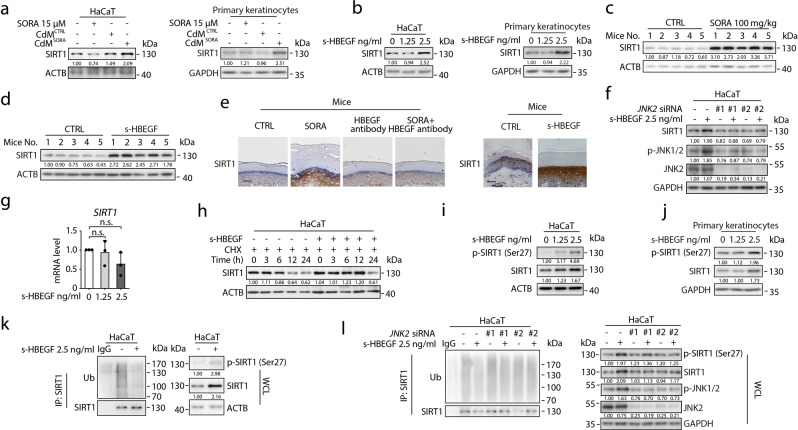
Hand-foot skin reaction (HFSR), among the most significant adverse effects of sorafenib, has been limiting the clinical benefits of this frontline drug in treating various malignant tumors. The mechanism underlying such toxicity remains poorly understood, hence the absence of effective intervention strategies. In the present study, we show that vascular endothelial cells are the primary cellular target of sorafenib-induced HFSR wherein soluble heparin-binding epidermal growth factor (s-HBEGF) mediates the crosstalk between vascular endothelial cells and keratinocytes. Mechanistically, s-HBEGF released from vascular endothelial cells activates the epidermal growth factor receptor (EGFR) on keratinocytes and promotes the phosphorylation of c-Jun N-terminal kinase 2 (JNK2), which stabilizes sirtuin 1 (SIRT1), an essential keratinization inducer, and ultimately gives rise to HFSR. The administration of s-HBEGF in vivo could sufficiently induce hyper-keratinization without sorafenib treatment. Furthermore, we report that HBEGF neutralization antibody, Sirt1 knockdown, and a classic SIRT1 inhibitor nicotinamide could all significantly reduce the sorafenib-induced HFSR in the mouse model. It is noteworthy that nicotinic acid, a prodrug of nicotinamide, could substantially reverse the sorafenib-induced HFSR in ten patients in a preliminary clinical study. Collectively, our findings reveal the mechanism of vascular endothelial cell-promoted keratinization in keratinocytes and provide a potentially promising therapeutic strategy for the treatment of sorafenib-induced HFSR.
手足皮肤反应(HFSR)是索拉非尼最显著的不良反应之一,限制了该药作为一线药物治疗各种恶性肿瘤的临床获益。这种毒性的机制仍不清楚,因此缺乏有效的干预策略。在本研究中,我们表明血管内皮细胞是索拉非尼诱导的 HFSR 的主要靶细胞,其中可溶性肝素结合表皮生长因子(s-HBEGF)介导血管内皮细胞和角质形成细胞之间的串扰。在机制上,血管内皮细胞释放的 s-HBEGF 激活角质形成细胞上的表皮生长因子受体(EGFR),并促进 c-Jun N 末端激酶 2(JNK2)的磷酸化,这稳定了丝氨酸/苏氨酸激酶 1(SIRT1),一种必不可少的角化诱导剂,最终导致 HFSR。体内给予 s-HBEGF 足以在没有索拉非尼治疗的情况下诱导过度角化。此外,我们报告说,HBEGF 中和抗体、Sirt1 敲低和经典 SIRT1 抑制剂烟酰胺都可以在小鼠模型中显著减少索拉非尼诱导的 HFSR。值得注意的是,烟酰胺的前体烟碱酸在初步临床研究中可以显著逆转 10 名患者的索拉非尼诱导的 HFSR。总之,我们的研究结果揭示了血管内皮细胞促进角质形成细胞角化的机制,并为治疗索拉非尼诱导的 HFSR 提供了一种有潜力的治疗策略。