Department of Computer Science, University of Illinois at Urbana-Champaign, 201 N Goodwin Ave, Urbana, 61801, IL, US.
BMC Genomics. 2020 Apr 16;21(Suppl 2):235. doi: 10.1186/s12864-020-6605-1.
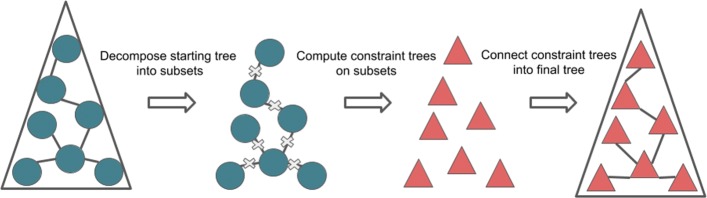
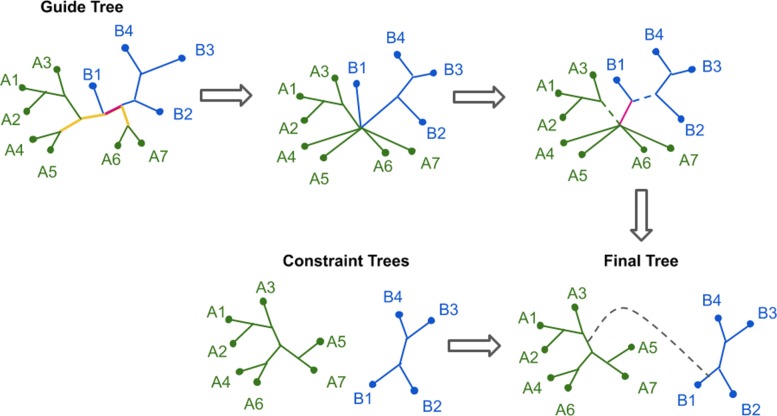
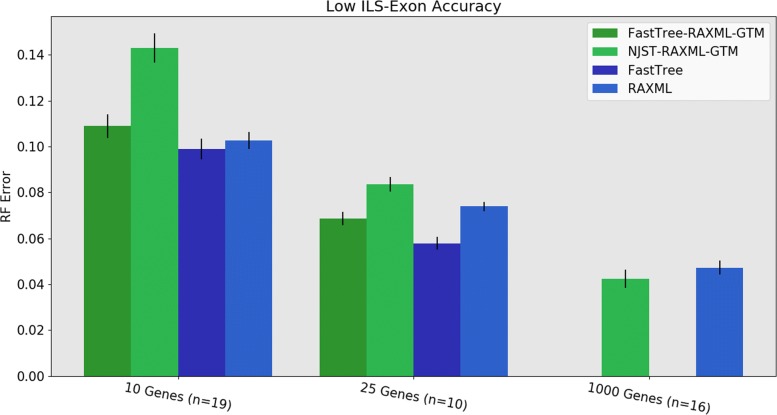
Phylogeny estimation is an important part of much biological research, but large-scale tree estimation is infeasible using standard methods due to computational issues. Recently, an approach to large-scale phylogeny has been proposed that divides a set of species into disjoint subsets, computes trees on the subsets, and then merges the trees together using a computed matrix of pairwise distances between the species. The novel component of these approaches is the last step: Disjoint Tree Merger (DTM) methods.
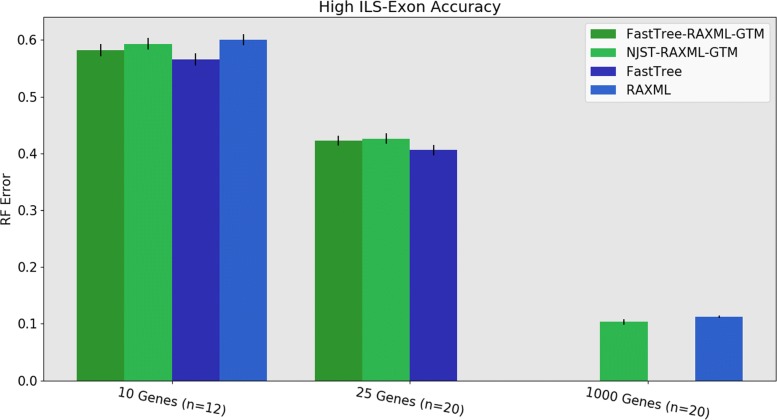
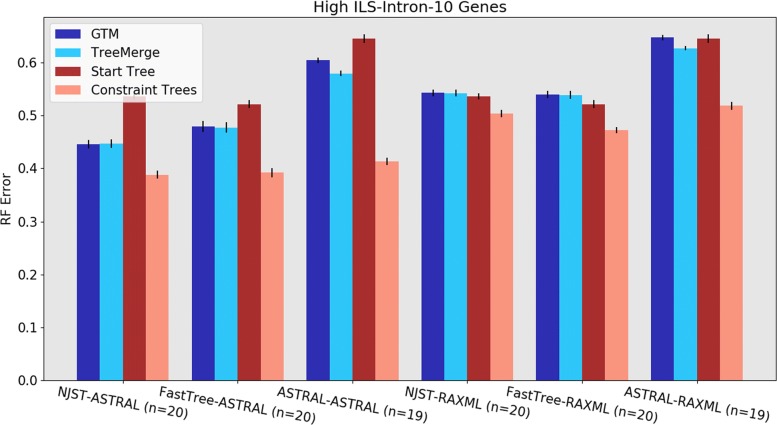
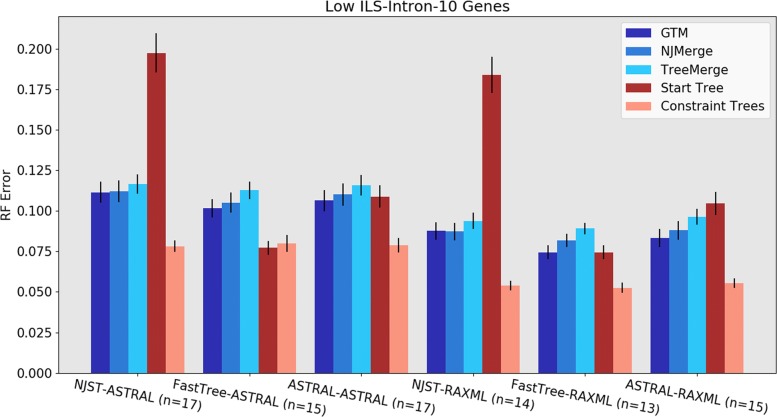
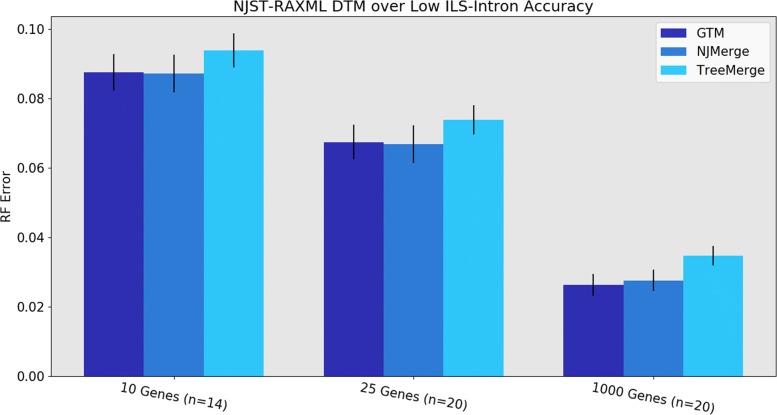
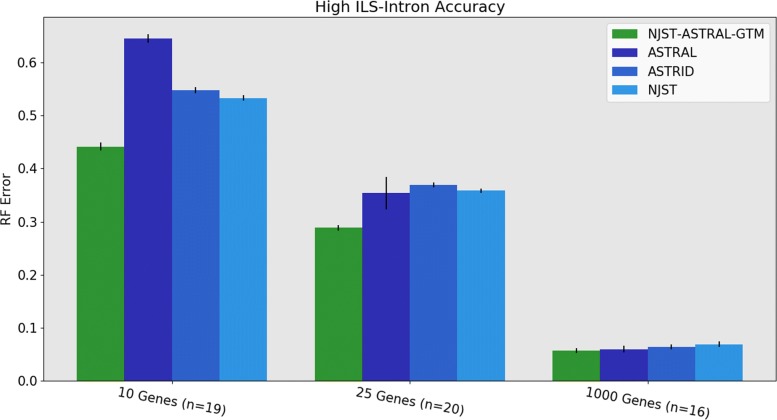
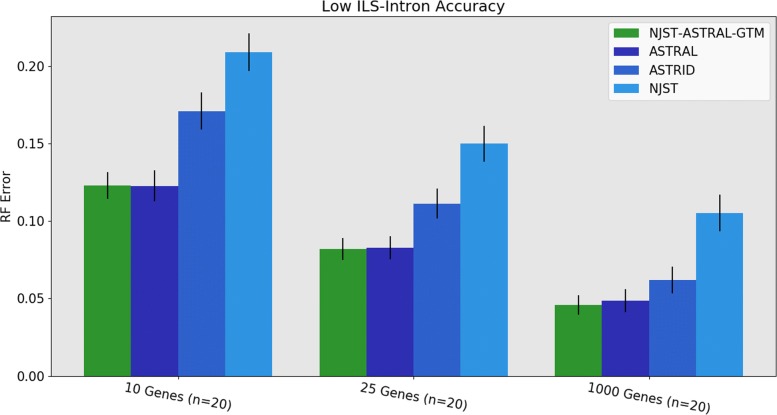
We present GTM (Guide Tree Merger), a polynomial time DTM method that adds edges to connect the subset trees, so as to provably minimize the topological distance to a computed guide tree. Thus, GTM performs unblended mergers, unlike the previous DTM methods. Yet, despite the potential limitation, our study shows that GTM has excellent accuracy, generally matching or improving on two previous DTMs, and is much faster than both.
The proposed GTM approach to the DTM problem is a useful new tool for large-scale phylogenomic analysis, and shows the surprising potential for unblended DTM methods.
系统发育估计是许多生物学研究的重要组成部分,但由于计算问题,使用标准方法进行大规模树估计是不可行的。最近,提出了一种大规模系统发育的方法,该方法将一组物种划分为不相交的子集,在子集中计算树,然后使用物种之间计算的成对距离矩阵将树合并在一起。这些方法的新颖之处在于最后一步:不相交树合并(DTM)方法。
我们提出了 GTM(引导树合并),这是一种多项式时间的 DTM 方法,它添加边来连接子集树,从而可证明地最小化到计算出的引导树的拓扑距离。因此,GTM 执行未混合的合并,与以前的 DTM 方法不同。然而,尽管存在潜在的局限性,但我们的研究表明,GTM 具有出色的准确性,通常与两个以前的 DTM 匹配或改进,并且比两者都快得多。
提出的 DTM 问题的 GTM 方法是大规模基因组分析的有用新工具,并显示出未混合 DTM 方法的惊人潜力。