Joint Department of Biomedical Engineering, University of North Carolina, and North Carolina State University, Chapel Hill and Raleigh, North Carolina, USA.
Department of Molecular Biomedical Sciences and Comparative Medicine Institute, North Carolina State University, Raleigh, North Carolina, USA.
Stem Cells Transl Med. 2020 Jul;9(7):786-798. doi: 10.1002/sctm.19-0167. Epub 2020 Apr 18.
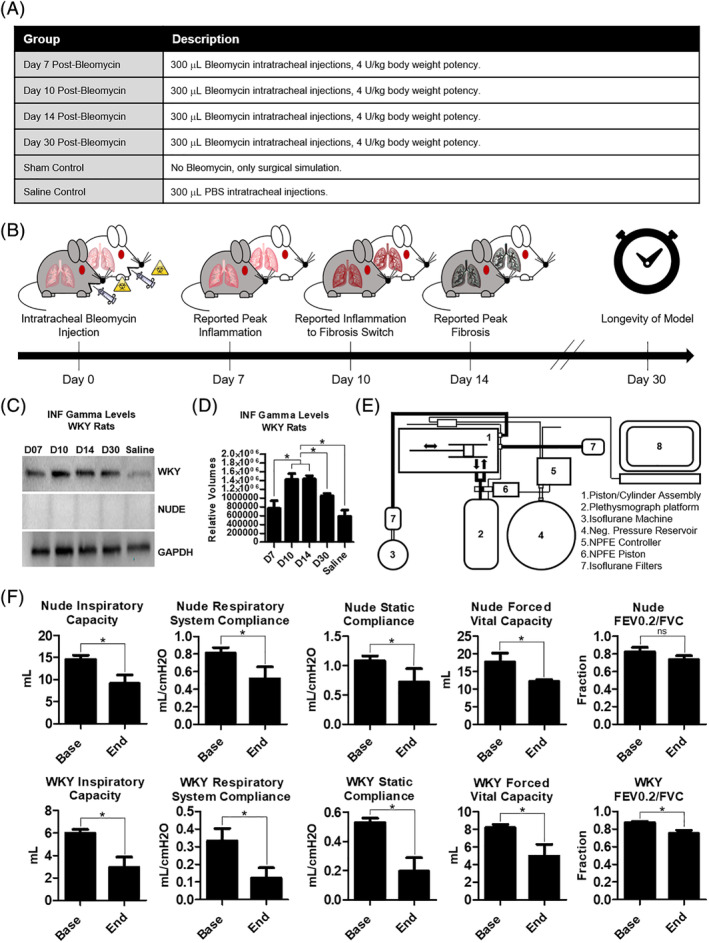
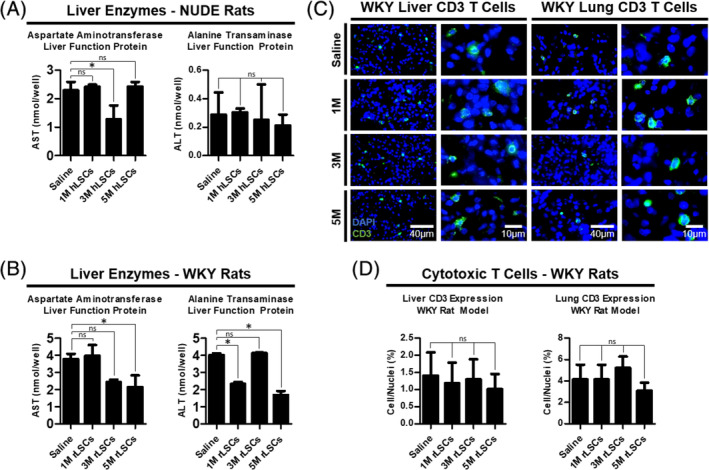
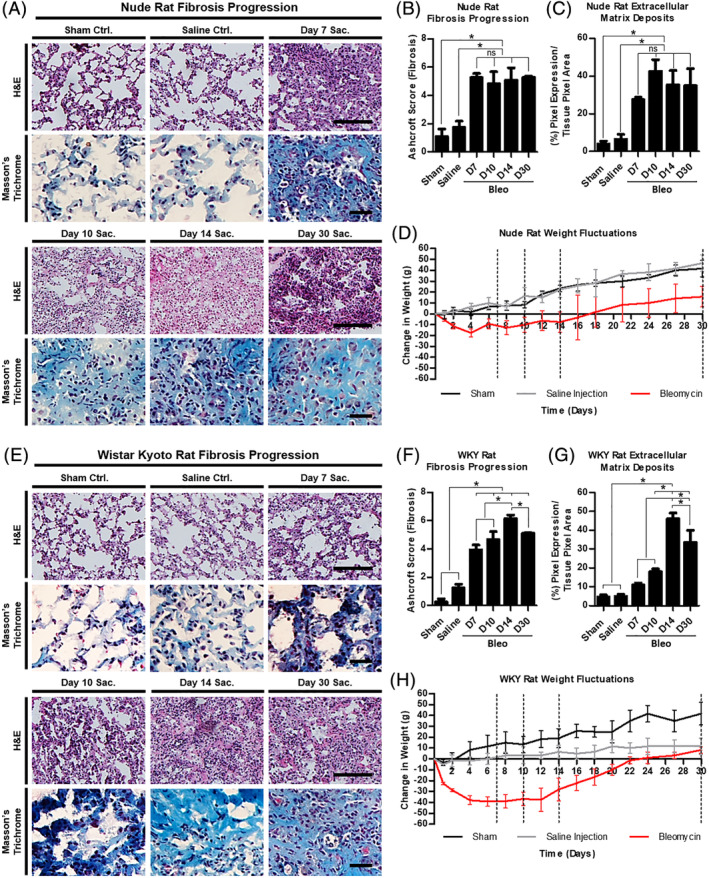
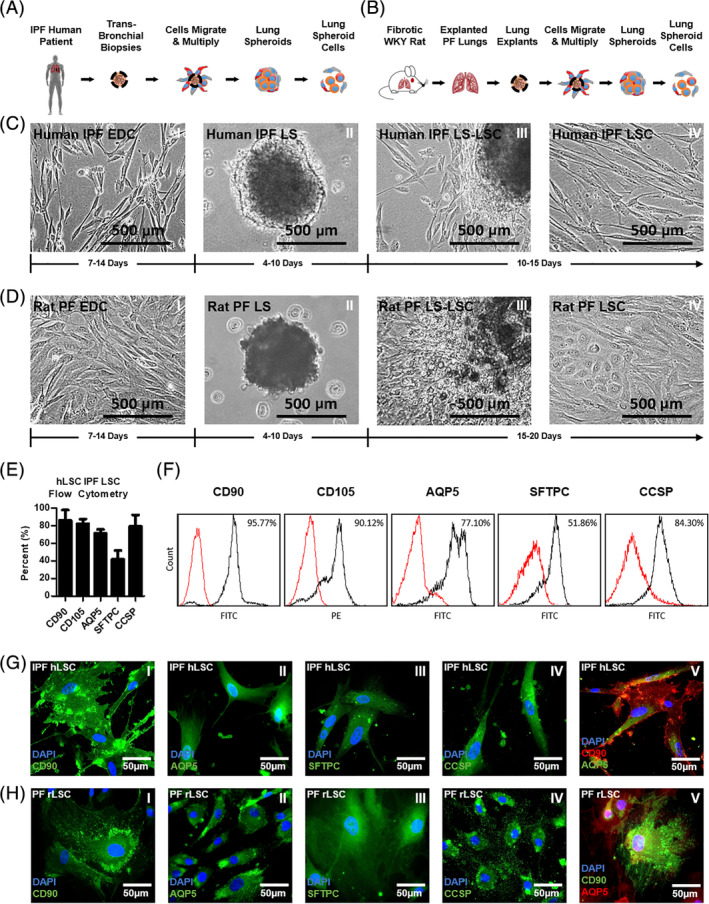
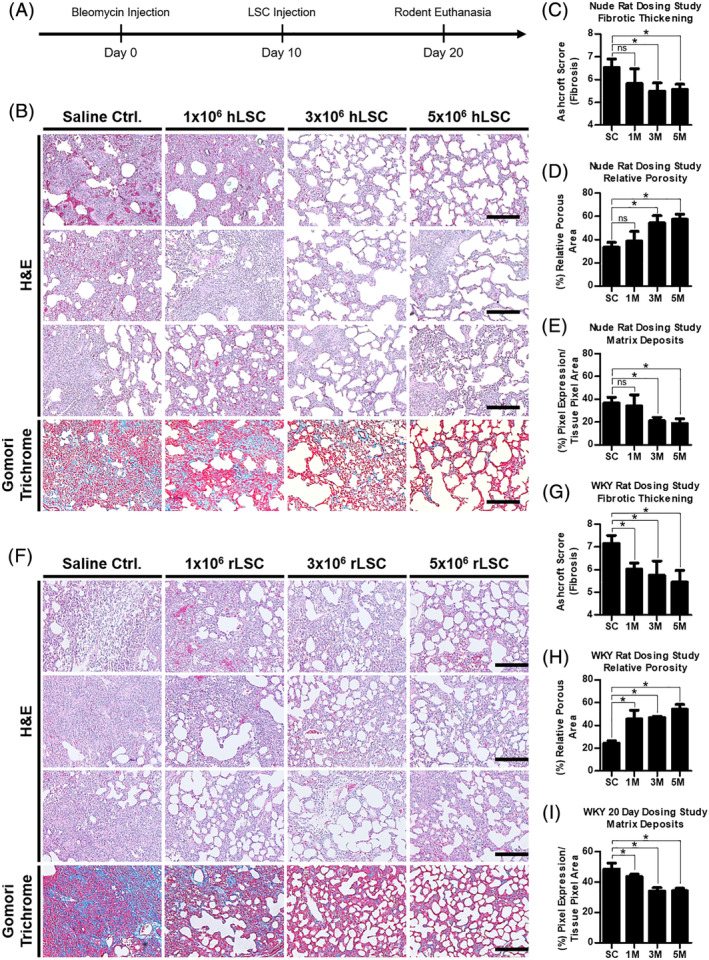
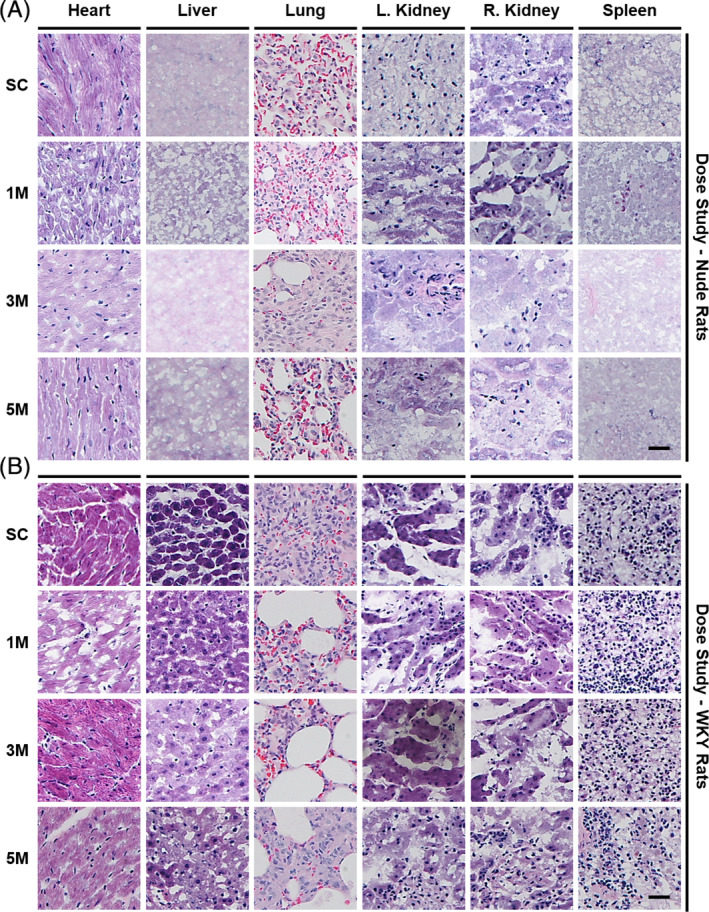
Idiopathic pulmonary fibrosis is a lethal interstitial lung disease with unknown etiology, no cure, and few treatment options. Herein, a therapy option is presented that makes use of a heterogeneous population of lung cells, including progenitor cells and supporting cells lines, cultured in adherent and suspension conditions, the latter of which induces spontaneous spheroid formation. Within these spheroids, progenitor marker expression is augmented. The cells, called lung spheroid cells, are isolated from fibrotic lungs, expanded, and delivered in single cell suspensions into rat models of pulmonary fibrosis via tail-vein injections. Two bleomycin-induced fibrotic rat models are used; a syngeneic Wistar-Kyoto rat model, treated with syngeneic cells, and a xenogeneic nude rat model, treated with human cells. The first objective was to study the differences in fibrotic progression in the two rat models after bleomycin injury. Nude rat fibrosis formed quickly and extended for 30 days with no self-resolution. Wistar-Kyoto rat fibrosis was more gradual and began to decrease in severity between days 14 and 30. The second goal was to find the minimum effective dose of cells that demonstrated safe and effective therapeutic value. The resultant minimum effective therapeutic dose, acquired from the nude rat model, was 3 × 10 human cells. Histological analysis revealed no evidence of tumorigenicity, increased local immunological activity in the lungs, or an increase in liver enzyme production. These data demonstrate the safety and efficacy of lung spheroid cells in their application as therapeutic agents for pulmonary fibrosis, as well as their potential for clinical translation.
特发性肺纤维化是一种病因不明、无法治愈且治疗选择有限的致命性间质性肺病。在此,提出了一种治疗选择,该选择利用了包括祖细胞和支持细胞系在内的异质肺细胞群体,在贴壁和悬浮条件下培养,后者诱导自发球体形成。在这些球体中,祖细胞标志物表达增强。这些细胞称为肺球体细胞,从纤维化肺中分离出来,扩增后以单细胞悬液形式通过尾静脉注射递送至肺纤维化大鼠模型中。使用了两种博来霉素诱导的纤维化大鼠模型;一种是同基因 Wistar-Kyoto 大鼠模型,用同基因细胞治疗;另一种是异种 nude 大鼠模型,用人细胞治疗。第一个目标是研究博来霉素损伤后两种大鼠模型中纤维化进展的差异。nude 大鼠的纤维化迅速形成并持续 30 天,没有自行缓解。Wistar-Kyoto 大鼠的纤维化则较为缓慢,在第 14 天至 30 天之间开始减轻。第二个目标是确定显示安全有效治疗价值的最小有效细胞剂量。从 nude 大鼠模型中获得的最小有效治疗剂量为 3×10 个人体细胞。组织学分析显示没有肿瘤发生的证据、肺部局部免疫活性增加或肝酶产生增加。这些数据证明了肺球体细胞作为肺纤维化治疗剂的安全性和有效性,以及它们在临床转化方面的潜力。