Alvarenga Amanda B, Veroneze Renata, Oliveira Hinayah R, Marques Daniele B D, Lopes Paulo S, Silva Fabyano F, Brito Luiz F
Department of Animal Sciences, Purdue University, West Lafayette, IN, United States.
Department of Animal Science, Federal University of Viçosa, Viçosa, Brazil.
Front Genet. 2020 Apr 9;11:263. doi: 10.3389/fgene.2020.00263. eCollection 2020.
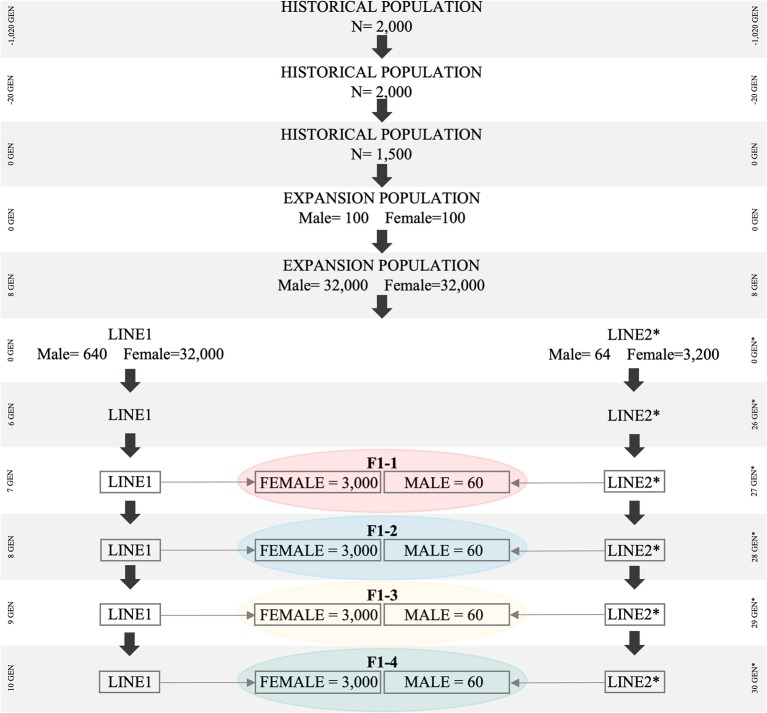
As crossbreeding is extensively used in some livestock species, we aimed to evaluate the performance of single-step GBLUP (ssGBLUP) and weighted ssGBLUP (WssGBLUP) methods to predict Genomic Estimated Breeding Values (GEBVs) of crossbred animals. Different training population scenarios were evaluated: (SC1) ssGBLUP based on a single-trait model considering purebred and crossbred animals in a joint training population; (SC2) ssGBLUP based on a multiple-trait model to enable considering phenotypes recorded in purebred and crossbred training animals as different traits; (SC3) WssGBLUP based on a single-trait model considering purebred and crossbred animals jointly in the training population (both populations were used for SNP weights' estimation); (SC4) WssGBLUP based on a single-trait model considering only purebred animals in the training population (crossbred population only used for SNP weights' estimation); (SC5) WssGBLUP based on a single-trait model and the training population characterized by purebred animals (purebred population used for SNP weights' estimation). A complex trait was simulated assuming alternative genetic architectures. Different scaling factors to blend the inverse of the genomic ( ) and pedigree ( ) relationship matrices were also tested. The predictive performance of each scenario was evaluated based on the validation accuracy and regression coefficient. The genetic correlations across simulated populations in the different scenarios ranged from moderate to high (0.71-0.99). The scenario mimicking a completely polygenic trait ( 0) yielded the lowest validation accuracy (0.12; for SC3 and SC4). The simulated scenarios assuming 4,500 QTLs affecting the trait and resulted in the greatest GEBV accuracies (0.47; for SC1 and SC2). The regression coefficients ranged from 0.28 (for SC3 assuming polygenic effect) to 1.27 (for SC2 considering 4,500 QTLs). In general, SC3 and SC5 resulted in inflated GEBVs, whereas other scenarios yielded deflated GEBVs. The scaling factors used to combine and had a small influence on the validation accuracies, but a greater effect on the regression coefficients. Due to the complexity of multiple-trait models and WssGBLUP analyses, and a similar predictive performance across the methods evaluated, SC1 is recommended for genomic evaluation in crossbred populations with similar genetic structures [moderate-to-high (0.71-0.99) genetic correlations between purebred and crossbred populations].
由于杂交在一些家畜品种中广泛应用,我们旨在评估单步基因组最佳线性无偏预测法(ssGBLUP)和加权单步基因组最佳线性无偏预测法(WssGBLUP)预测杂交动物基因组估计育种值(GEBVs)的性能。评估了不同的训练群体方案:(SC1)基于单性状模型的ssGBLUP,在联合训练群体中考虑纯种和杂交动物;(SC2)基于多性状模型的ssGBLUP,以便将纯种和杂交训练动物记录的表型视为不同性状;(SC3)基于单性状模型的WssGBLUP,在训练群体中联合考虑纯种和杂交动物(两个群体均用于SNP权重估计);(SC4)基于单性状模型的WssGBLUP,训练群体中仅考虑纯种动物(杂交群体仅用于SNP权重估计);(SC5)基于单性状模型的WssGBLUP,训练群体以纯种动物为特征(纯种群体用于SNP权重估计)。假设不同的遗传结构模拟了一个复杂性状。还测试了用于混合基因组( )和系谱( )关系矩阵逆矩阵的不同缩放因子。基于验证准确性和回归系数评估了每种方案的预测性能。不同方案中模拟群体间的遗传相关性从中等到高度(0.71 - 0.99)。模拟完全多基因性状( 0)的方案产生了最低的验证准确性(0.12;适用于SC3和SC4)。假设影响该性状的有4500个数量性状位点(QTLs)的模拟方案导致了最高的GEBV准确性(0.47;适用于SC1和SC2)。回归系数范围从0.28(对于假设多基因效应的SC3)到1.27(对于考虑4500个QTLs的SC2)。总体而言,SC3和SC5导致GEBVs膨胀,而其他方案导致GEBVs缩减。用于组合 和 的缩放因子对验证准确性影响较小,但对回归系数影响较大。由于多性状模型和WssGBLUP分析的复杂性,以及所评估方法间相似的预测性能,对于具有相似遗传结构[纯种和杂交群体间中等至高(0.71 - 0.99)遗传相关性]的杂交群体基因组评估,建议采用SC1。