Key Laboratory of Pollinating Insect Biology, Ministry of Agriculture; Institute of Apicultural Research, Chinese Academy of Agricultural Sciences, 100093, Beijing, China.
College of Agriculture and Environmental Sciences, Bahir Dar University, Bahir Dar, Ethiopia.
Sci Rep. 2020 May 5;10(1):7532. doi: 10.1038/s41598-020-64022-3.
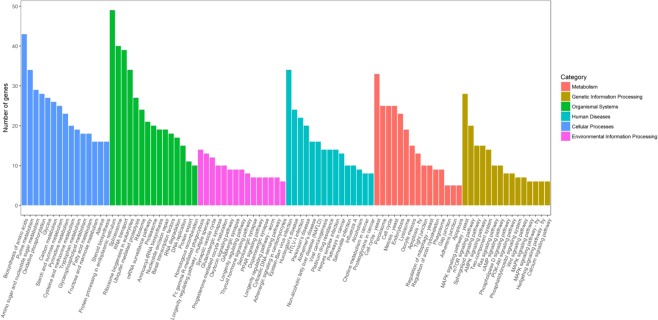
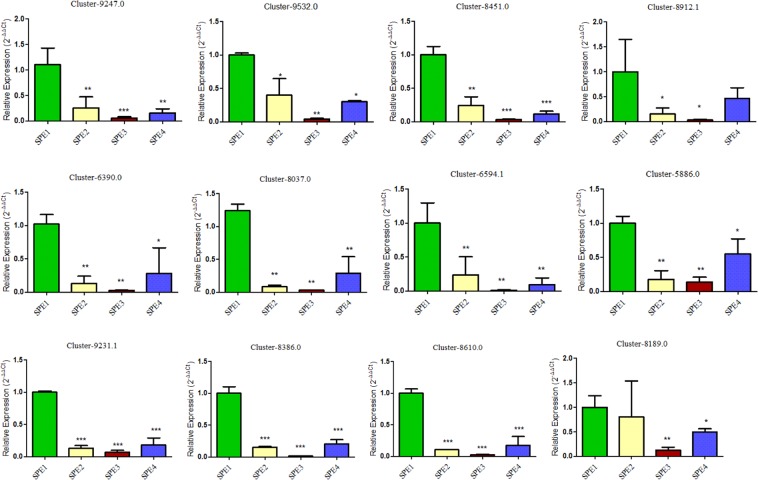
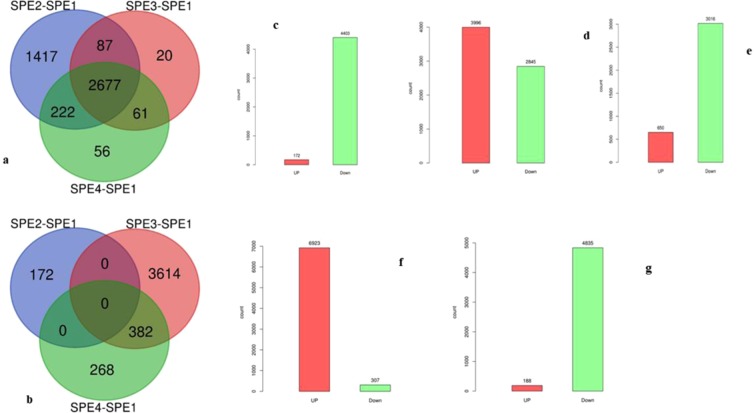
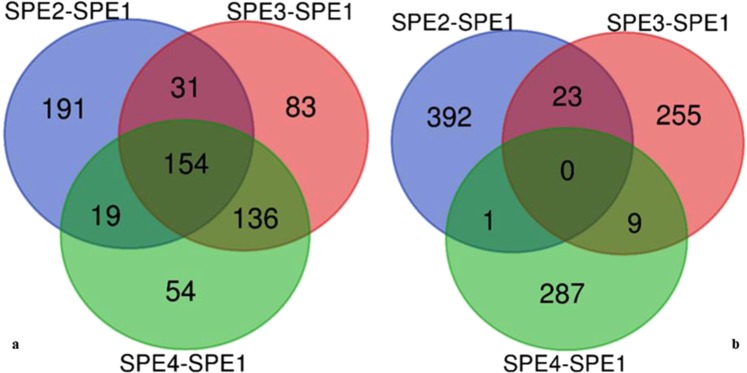
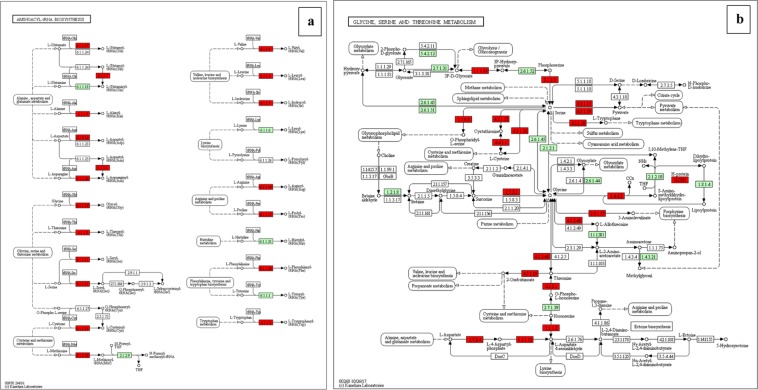
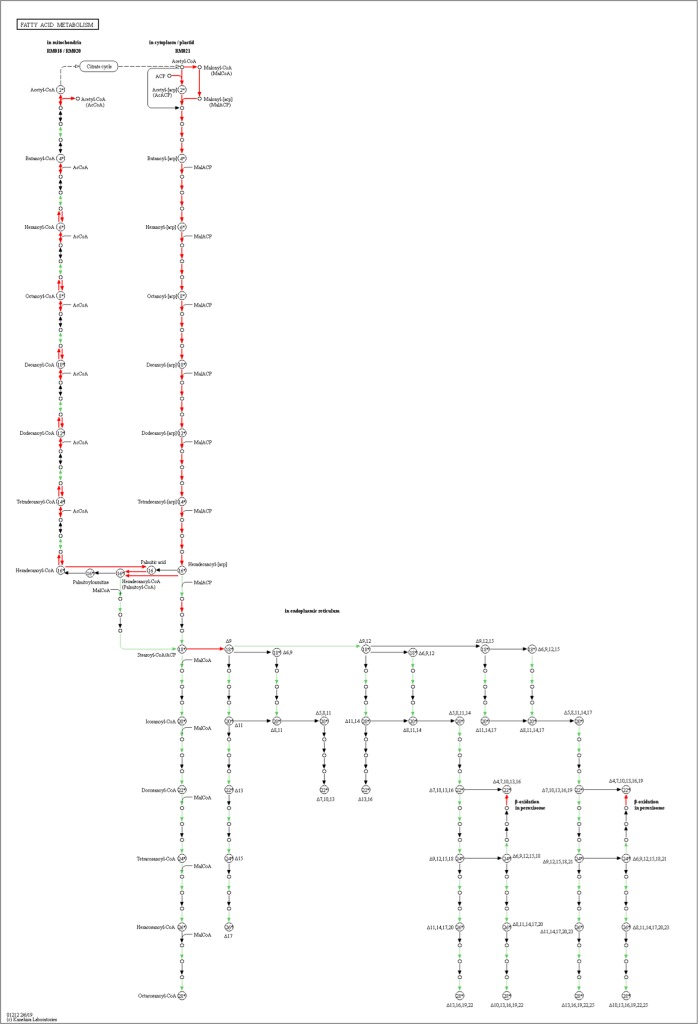
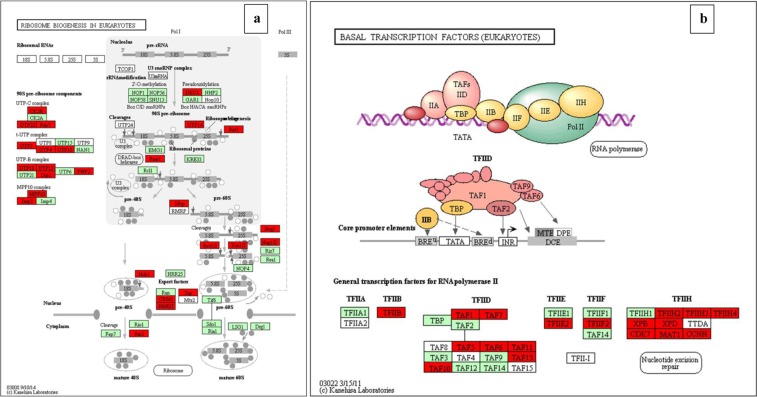
Chalkbrood disease is caused by Ascosphaera apis which severely affects honeybee brood. Spore inoculation experiments shown pathogenicity varies among different strains and mutants, however, the molecular mechanism of pathogenicity is unclear. We sequenced, assembled and annotated the transcriptomes of wild type (SPE1) and three mutants (SPE2, SPE3 and SPE4) with reduced pathogenicity that were constructed in our previous study. Illumina sequencing generated a total of 394,910,604 clean reads and de novo Trinity-based assembled into 12,989 unigenes, among these, 9,598 genes were successfully annotated to known proteins in UniProt database. A total of 172, 3,996, and 650 genes were up-regulated and 4,403, 2,845, and 3,016 genes were down-regulated between SPE2-SPE1, SPE3-SPE1, and SPE4-SPE1, respectively. Overall, several genes with a potential role in fungal pathogenicity were detected down-regulated in mutants including 100 hydrolytic enzymes, 117 transcriptional factors, and 47 cell wall related genes. KEGG pathway enrichment analysis reveals 216 genes involved in nine pathways were down-regulated in mutants compared to wild type. The down-regulation of more pathways involved in pathogenicity in SPE2 and SPE4 than SPE3 supports their lower pathogenicity during in-vitro bioassay experiment. Expression of 12 down-regulated genes in mutants was validated by quantitative real time PCR. This study provides valuable information on transcriptome variation caused by mutation for further functional validation of candidate pathogenicity genes in A. apis.
chalkbrood 病是由 Ascosphaera apis 引起的,严重影响蜜蜂幼虫。孢子接种实验表明,不同菌株和突变体的致病性存在差异,但致病性的分子机制尚不清楚。我们对之前研究中构建的具有较低致病性的野生型(SPE1)和 3 个突变体(SPE2、SPE3 和 SPE4)的转录组进行了测序、组装和注释。Illumina 测序共产生了 394,910,604 条清洁reads,基于 Trinity 的 de novo 组装成 12,989 条 unigenes,其中 9,598 条基因成功注释到 UniProt 数据库中的已知蛋白。SPE2-SPE1、SPE3-SPE1 和 SPE4-SPE1 之间分别有 172、3,996 和 650 个基因上调,4,403、2,845 和 3,016 个基因下调。总的来说,在突变体中检测到一些可能与真菌致病性相关的基因下调,包括 100 种水解酶、117 种转录因子和 47 种细胞壁相关基因。KEGG 通路富集分析显示,与野生型相比,突变体中有 216 个基因参与的 9 条通路下调。SPE2 和 SPE4 比 SPE3 中更多与致病性相关的通路下调,支持它们在体外生物测定实验中较低的致病性。通过定量实时 PCR 验证了 12 个突变体中下调基因的表达。本研究为突变引起的转录组变化提供了有价值的信息,为进一步验证 A. apis 中候选致病性基因的功能提供了依据。