Harvard Reproductive Endocrine Sciences Center, Reproductive Endocrine Unit of the Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts 02114, USA.
Newcastle-upon-Tyne Hospitals Foundation NHS Trust (Royal Victoria Infirmary) and Institute of Genetic Medicine, University of Newcastle-upon-Tyne, Newcastle-upon-Tyne NE1 7RU, United Kingdom.
Cold Spring Harb Mol Case Stud. 2020 Jun 12;6(3). doi: 10.1101/mcs.a005033. Print 2020 Jun.
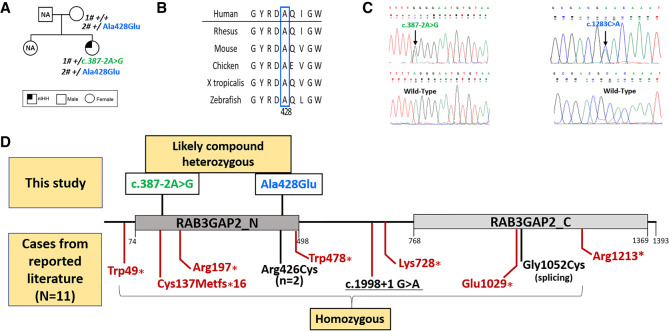
Biallelic pathogenic variants in cause Warburg Micro syndrome (WARBM) and Martsolf syndrome (MS), two rare, phenotypically overlapping disorders characterized by congenital cataracts, intellectual disability, and hypogonadism. Although the initial report documented hypergonadotropic hypogonadism (implying a gonadal defect), an adolescent girl with WARBM/MS was subsequently reported to have hypogonadotropic hypogonadism (implying a central defect in either the hypothalamus or anterior pituitary). However, in adult MS, hypogonadotropism has not been convincingly demonstrated. Additionally, the correlation between the pathogenic severity of variants in and the phenotypic severity also remains unclear. Here we present a clinical report of a woman with congenital cataracts, apparent intellectual disability, and pubertal failure who underwent exome sequencing (ES) to determine a precise molecular diagnosis. Reproductive phenotypes reported previously in individuals with MS and the genotypic spectrum of previous variants were also reviewed. The ES identified pathogenic compound heterozygous variants (c.387-2A > G; p.(Arg428Glu)) combined with her phenotypic features, which enabled a unifying molecular diagnosis of MS. Reproductive evaluation confirmed a normosmic idiopathic hypogonadotropic hypogonadism. Review of the allelic spectrum in WARBM/MS suggests that although variants resulting in complete abrogation of RAB3GAP2 protein function cause severe WARBM, variants associated with partially preserved RAB3GAP2 function cause milder MS. This report expands the genotypic and phenotypic spectrum of MS and demonstrates hypogonadotropic hypogonadism as a key pathophysiologic abnormality in MS. Genotype-phenotype associations of previously reported variants indicate that variants that fully abolish RAB3GAP2 function result in WARBM, whereas MS is associated with variants of lesser severity with residual RAB3GAP2 function.
在 中发现的双等位致病性变异导致沃伯格微综合征 (WARBM) 和马索夫综合征 (MS),这两种罕见的疾病表现重叠,其特征为先天性白内障、智力障碍和性腺功能减退。尽管最初的报告记录了促性腺激素性性腺功能减退症(意味着性腺缺陷),但随后报道了一名患有 WARBM/MS 的青春期女孩患有促性腺激素释放激素缺乏性性腺功能减退症(意味着下丘脑或垂体前叶存在中枢缺陷)。然而,在成人 MS 中,促性腺激素缺乏症尚未得到令人信服的证明。此外,在 中变异的致病性严重程度与表型严重程度之间的相关性也不清楚。在这里,我们报告了一名患有先天性白内障、明显智力障碍和青春期发育失败的女性的临床病例,她接受了外显子组测序 (ES) 以确定精确的分子诊断。还回顾了之前在 MS 患者中报告的生殖表型和之前 变异的基因型谱。ES 确定了致病性复合杂合的 变异(c.387-2A > G;p.(Arg428Glu)),结合其表型特征,可对 MS 进行统一的分子诊断。生殖评估证实为正常嗅觉特发性促性腺激素释放激素缺乏性性腺功能减退症。对 WARBM/MS 中 等位基因谱的回顾表明,尽管导致 RAB3GAP2 蛋白功能完全缺失的变异导致严重的 WARBM,但与部分保留 RAB3GAP2 功能相关的变异导致较轻的 MS。本报告扩展了 MS 的基因型和表型谱,并证明了促性腺激素释放激素缺乏性性腺功能减退症是 MS 的关键病理生理异常。以前报道的 变异的基因型-表型关联表明,完全消除 RAB3GAP2 功能的变异导致 WARBM,而 MS 与具有残余 RAB3GAP2 功能的较轻变异相关。