Department of Human Genetics, Leiden University Medical Centre, Leiden, The Netherlands.
Sequence Analysis Support Core, Leiden University Medical Centre, Leiden, The Netherlands.
Int J Cancer. 2020 Nov 15;147(10):2708-2716. doi: 10.1002/ijc.33039. Epub 2020 May 30.
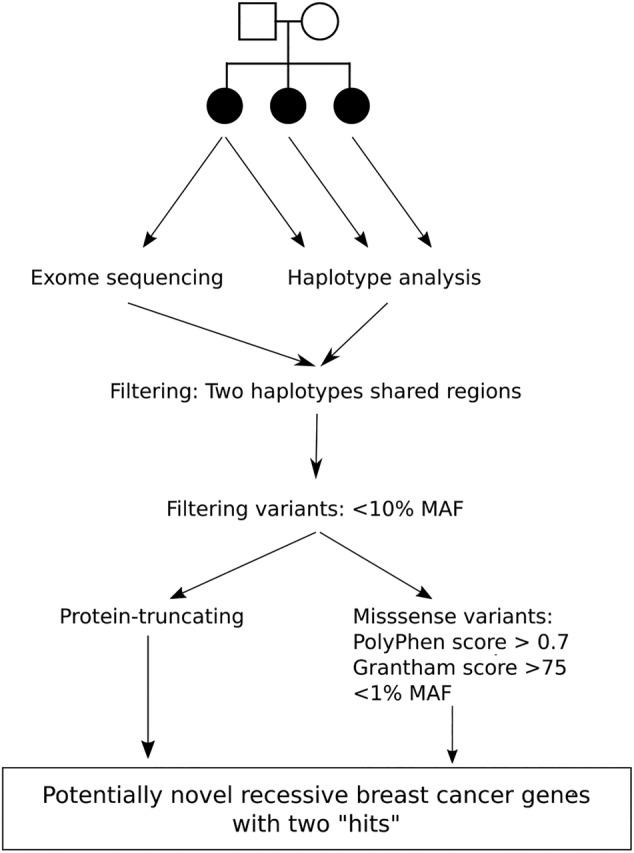
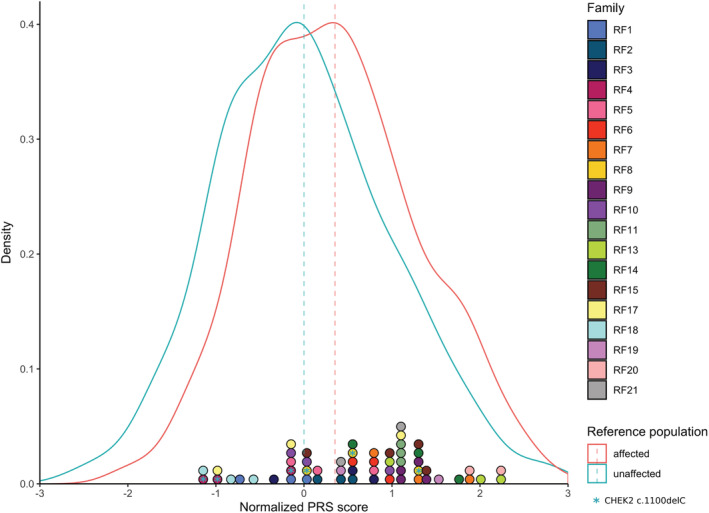
Breast cancer risk is approximately twice as high in first-degree relatives of female breast cancer cases than in women in the general population. Less than half of this risk can be attributed to the currently known genetic risk factors. Recessive risk alleles represent a relatively underexplored explanation for the remainder of familial risk. To address this, we selected 19 non-BRCA1/2 breast cancer families in which at least three siblings were affected, while no first-degree relatives of the previous or following generation had breast cancer. Germline DNA from one of the siblings was subjected to exome sequencing, while all affected siblings were genotyped using SNP arrays to assess haplotype sharing and to calculate a polygenic risk score (PRS) based on 160 low-risk variants. We found no convincing candidate recessive alleles among exome sequencing variants in genomic regions for which all three siblings shared two haplotypes. However, we found two families in which all affected siblings carried the CHEK2*1100delC. In addition, the average normalized PRS of the "recessive" family probands (0.81) was significantly higher than that in both general population cases (0.35, P = .026) and controls (P = .0004). These findings suggest that the familial aggregation is, at least in part, explained by a polygenic effect of common low-risk variants and rarer intermediate-risk variants, while we did not find evidence of a role for novel recessive risk alleles.
乳腺癌风险在女性乳腺癌病例的一级亲属中约为普通人群的两倍。不到一半的风险可以归因于目前已知的遗传风险因素。隐性风险等位基因代表了家族风险剩余部分相对未被充分探索的解释。为了解决这个问题,我们选择了 19 个非 BRCA1/2 乳腺癌家族,其中至少有三个兄弟姐妹受到影响,而前一代或后一代的一级亲属没有乳腺癌。一个兄弟姐妹的种系 DNA 进行了外显子组测序,而所有受影响的兄弟姐妹都使用 SNP 芯片进行了基因分型,以评估单倍型共享并基于 160 个低风险变体计算多基因风险评分 (PRS)。我们在所有三个兄弟姐妹共享两个单倍型的基因组区域的外显子组测序变体中没有发现令人信服的候选隐性等位基因。然而,我们发现了两个家族,所有受影响的兄弟姐妹都携带 CHEK2*1100delC。此外,“隐性”家族先证者的平均归一化 PRS(0.81)明显高于普通人群病例(0.35,P =.026)和对照组(P =.0004)。这些发现表明,家族聚集至少部分是由常见低风险变体和罕见中等风险变体的多基因效应引起的,而我们没有发现新的隐性风险等位基因作用的证据。