Pagliarani Serena, Lucchiari Sabrina, Scarlato Marina, Redaelli Elisa, Modoni Anna, Magri Francesca, Fossati Barbara, Previtali Stefano C, Sansone Valeria A, Lecchi Marzia, Lo Monaco Mauro, Meola Giovanni, Comi Giacomo P
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy.
Department of Neurology and INSPE, IRCCS Ospedale San Raffaele, Milan, Italy.
Front Neurol. 2020 Apr 29;11:255. doi: 10.3389/fneur.2020.00255. eCollection 2020.
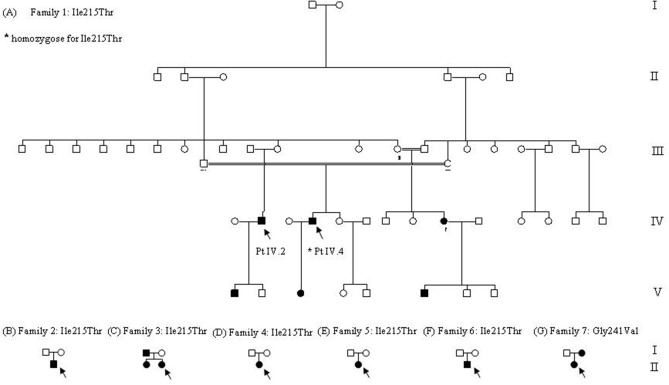
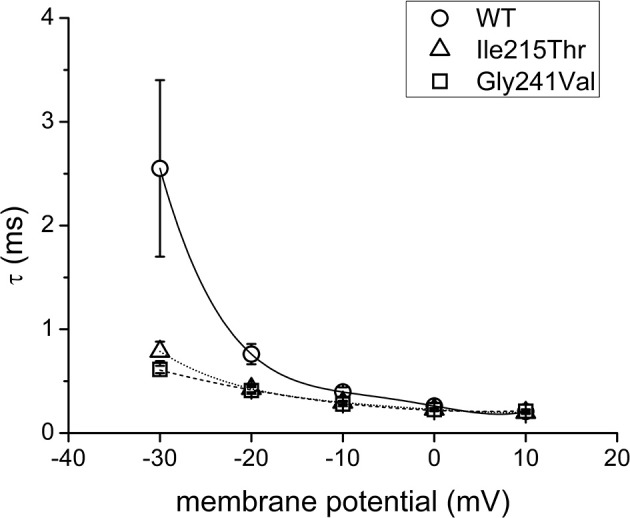
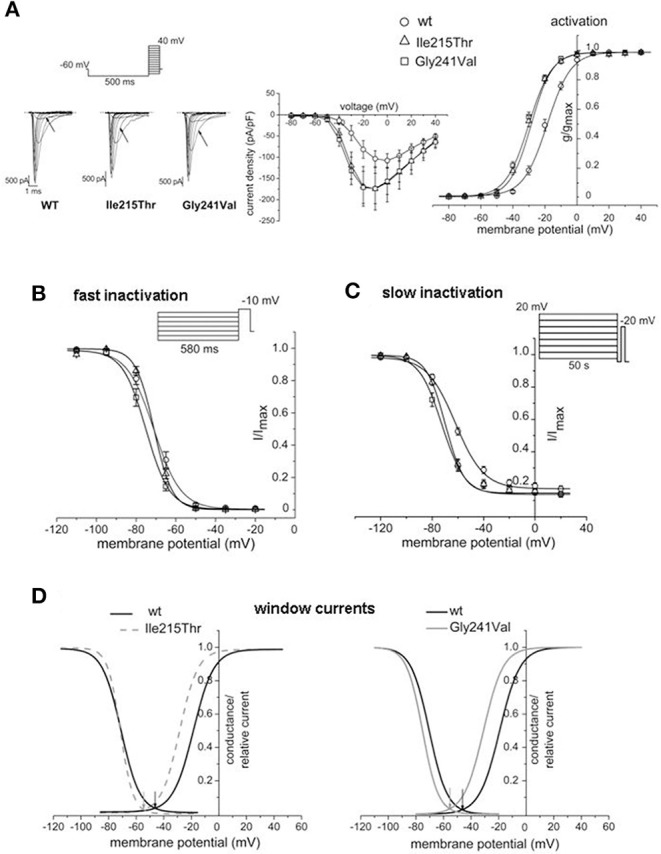
Sodium channel myotonia is a form of muscle channelopathy due to mutations that affect the Na1.4 channel. We describe seven families with a series of symptoms ranging from asymptomatic to clearly myotonic signs that have in common two novel mutations, p.Ile215Thr and p.Gly241Val, in the first domain of the Na1.4 channel. The families described have been clinically and genetically evaluated. p.Ile215Thr and p.Gly241Val lie, respectively, on extracellular and intracellular loops of the first domain of the Na1.4 channel. We assessed that the p.Ile215Thr mutation can be related to a founder effect in people from Southern Italy. Electrophysiological evaluation of the channel function showed that the voltage dependence of the activation for both the mutant channels was significantly shifted toward hyperpolarized potentials (Ile215Thr: -28.6 ± 1.5 mV and Gly241Val: -30.2 ± 1.3 mV vs. WT: -18.5 ± 1.3 mV). The slow inactivation was also significantly affected, whereas fast inactivation showed a different behavior in the two mutants. We characterized two novel mutations of the gene expanding the knowledge about genetics of mild forms of myotonia, and we present, to our knowledge, the first homozygous patient with sodium channel myotonia.
钠通道性肌强直是一种由于影响Na1.4通道的突变而导致的肌肉通道病。我们描述了七个家族,这些家族具有一系列症状,从无症状到明显的肌强直体征,它们在Na1.4通道的第一个结构域中共有两个新的突变,即p.Ile215Thr和p.Gly241Val。所描述的家族已经进行了临床和基因评估。p.Ile215Thr和p.Gly241Val分别位于Na1.4通道第一个结构域的细胞外环和细胞内环上。我们评估p.Ile215Thr突变可能与意大利南部人群中的奠基者效应有关。对通道功能的电生理评估表明,两种突变通道激活的电压依赖性均显著向超极化电位偏移(Ile215Thr:-28.6±1.5 mV,Gly241Val:-30.2±1.3 mV,而野生型为-18.5±1.3 mV)。慢失活也受到显著影响,而快失活在两种突变体中表现出不同的行为。我们鉴定了该基因的两个新突变,扩展了对轻度肌强直遗传学的认识,并且据我们所知,我们展示了首例钠通道性肌强直的纯合子患者。