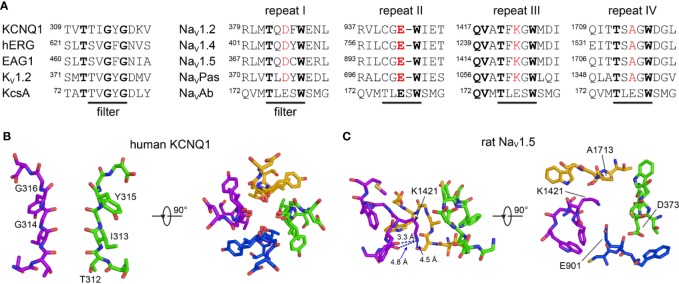
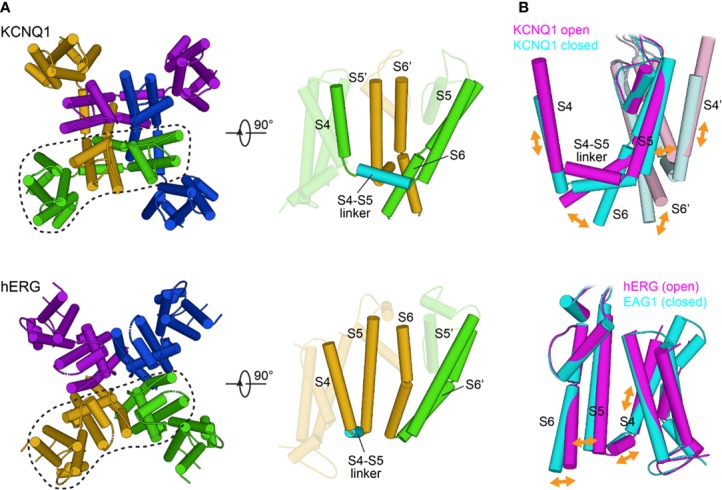
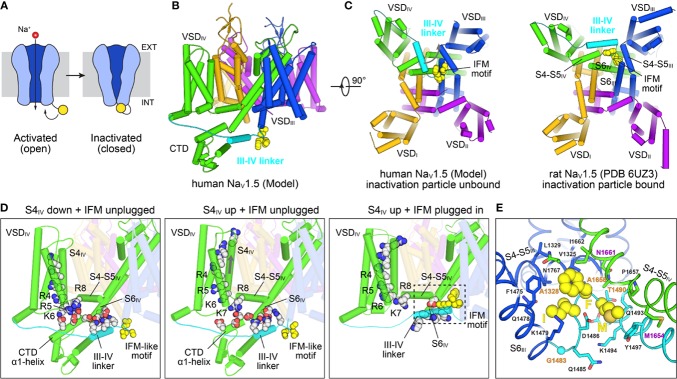
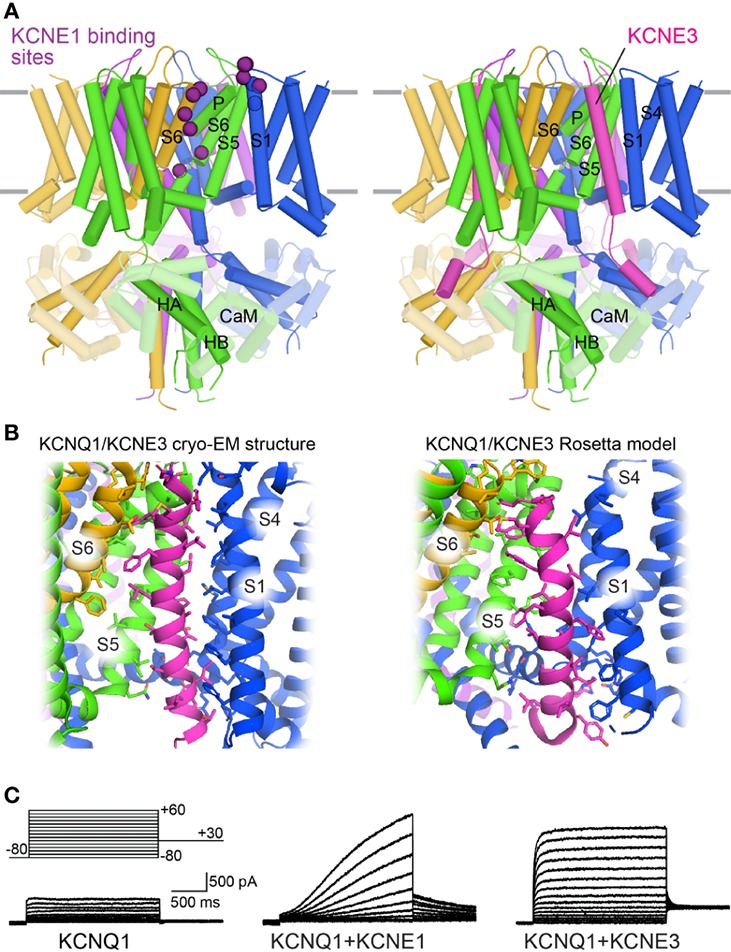
Brewer Kathryn R, Kuenze Georg, Vanoye Carlos G, George Alfred L, Meiler Jens, Sanders Charles R
Center for Structural Biology, Vanderbilt University School of Medicine Basic Sciences, Nashville, TN, United States.
Department of Biochemistry, Vanderbilt University, Nashville, TN, United States.
Front Pharmacol. 2020 May 4;11:550. doi: 10.3389/fphar.2020.00550. eCollection 2020.
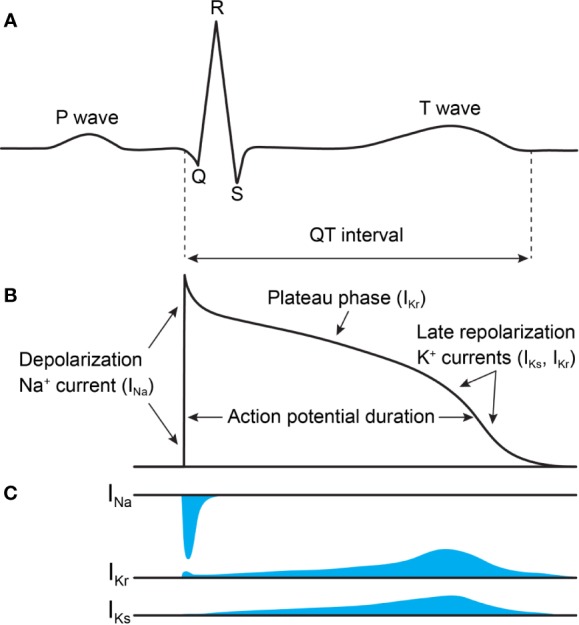
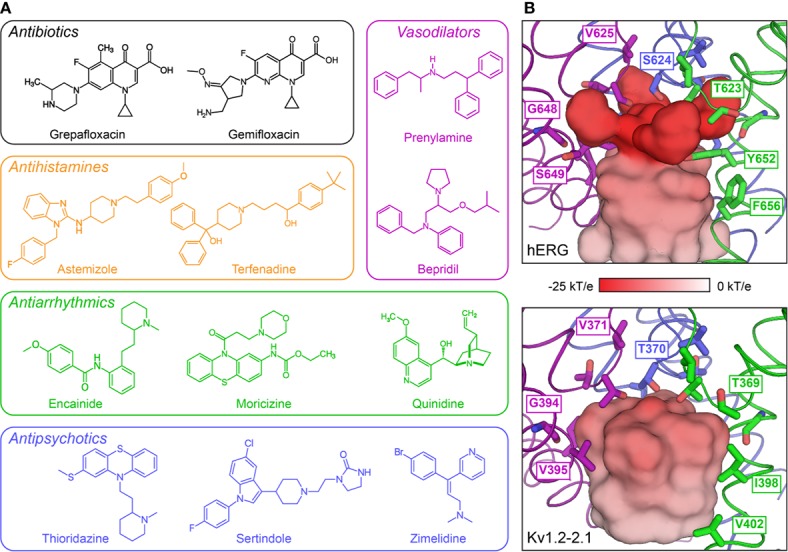
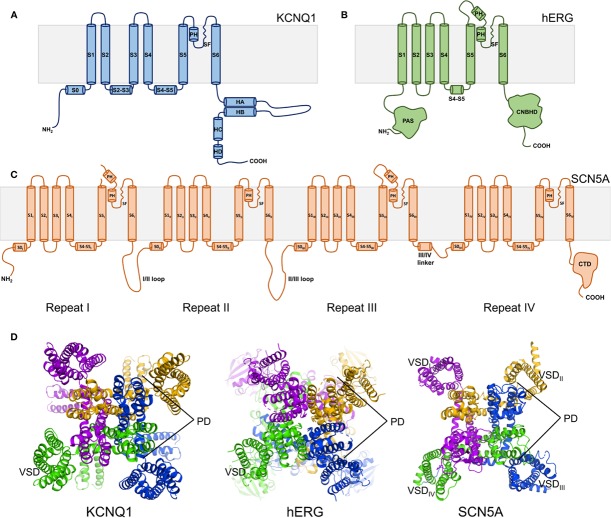
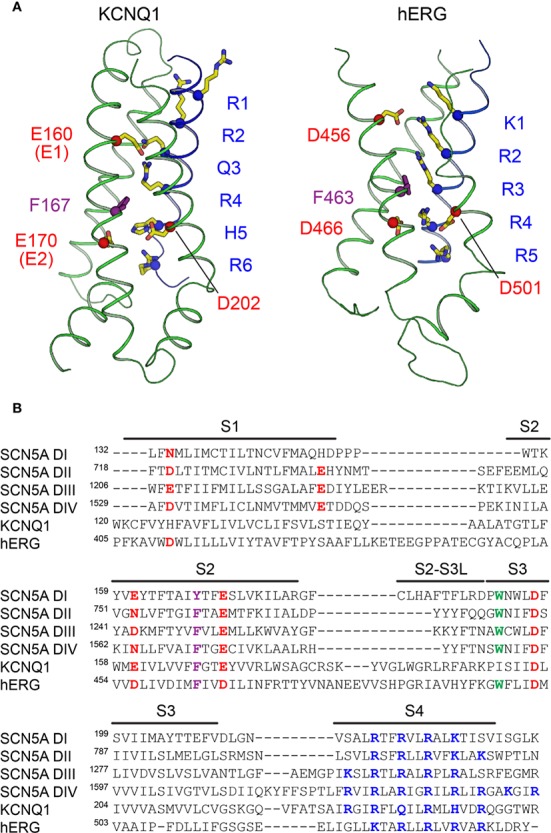
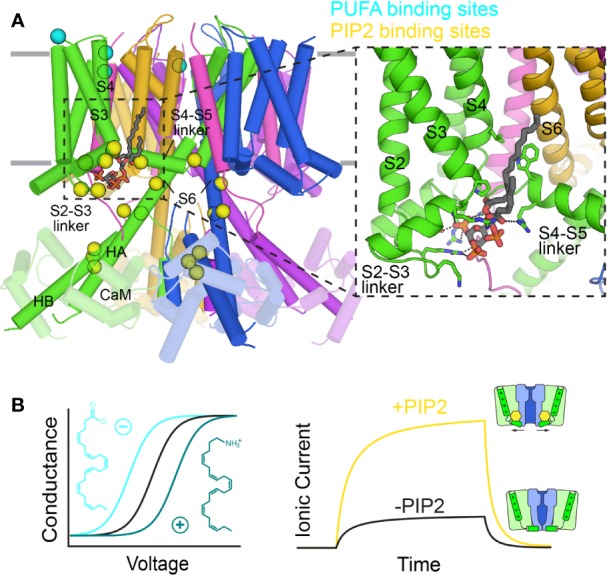
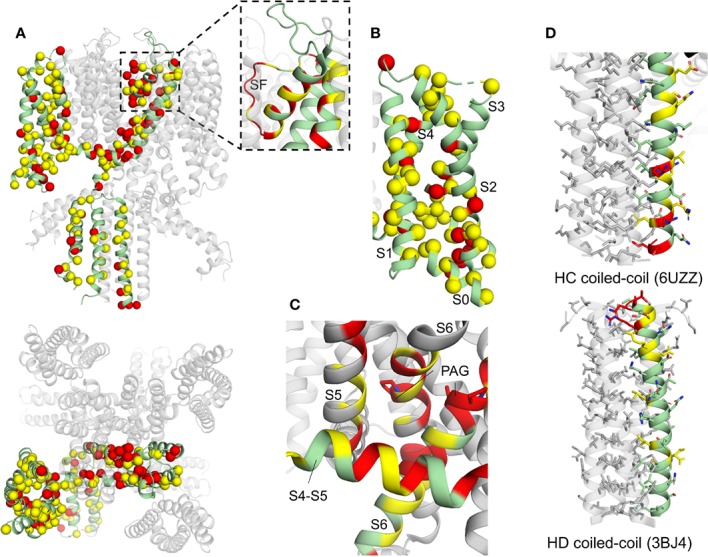
The cardiac action potential is critical to the production of a synchronized heartbeat. This electrical impulse is governed by the intricate activity of cardiac ion channels, among them the cardiac voltage-gated potassium (K) channels KCNQ1 and hERG as well as the voltage-gated sodium (Na) channel encoded by . Each channel performs a highly distinct function, despite sharing a common topology and structural components. These three channels are also the primary proteins mutated in congenital long QT syndrome (LQTS), a genetic condition that predisposes to cardiac arrhythmia and sudden cardiac death due to impaired repolarization of the action potential and has a particular proclivity for reentrant ventricular arrhythmias. Recent cryo-electron microscopy structures of human KCNQ1 and hERG, along with the rat homolog of SCN5A and other mammalian sodium channels, provide atomic-level insight into the structure and function of these proteins that advance our understanding of their distinct functions in the cardiac action potential, as well as the molecular basis of LQTS. In this review, the gating, regulation, LQTS mechanisms, and pharmacological properties of KCNQ1, hERG, and SCN5A are discussed in light of these recent structural findings.
心脏动作电位对于产生同步心跳至关重要。这种电冲动受心脏离子通道复杂活动的支配,其中包括心脏电压门控钾(K)通道KCNQ1和hERG,以及由[此处原文缺失相关基因名称]编码的电压门控钠(Na)通道。尽管这些通道具有共同的拓扑结构和结构成分,但每个通道都执行着高度独特的功能。这三种通道也是先天性长QT综合征(LQTS)中发生突变的主要蛋白质,LQTS是一种遗传性疾病,由于动作电位复极化受损,易导致心律失常和心源性猝死,并且特别容易发生折返性室性心律失常。最近人类KCNQ1和hERG的冷冻电子显微镜结构,以及SCN5A的大鼠同源物和其他哺乳动物钠通道的结构,为这些蛋白质的结构和功能提供了原子水平的见解,增进了我们对它们在心脏动作电位中不同功能的理解,以及对LQTS分子基础的理解。在这篇综述中,将根据这些最新的结构发现,讨论KCNQ1、hERG和SCN5A的门控、调节、LQTS机制和药理学特性。