Ammazzalorso Alessandra, Bruno Isabella, Florio Rosalba, De Lellis Laura, Laghezza Antonio, Cerchia Carmen, De Filippis Barbara, Fantacuzzi Marialuigia, Giampietro Letizia, Maccallini Cristina, Tortorella Paolo, Veschi Serena, Loiodice Fulvio, Lavecchia Antonio, Cama Alessandro, Amoroso Rosa
Department of Pharmacy, "G. d'Annunzio" University of Chieti-Pescara, Via Dei Vestini 31, 66100 Chieti, Italy.
Department of Pharmacy-Drug Science, University of Bari "Aldo Moro", Via E. Orabona 4, 70126 Bari, Italy.
ACS Med Chem Lett. 2020 Mar 3;11(5):624-632. doi: 10.1021/acsmedchemlett.9b00666. eCollection 2020 May 14.
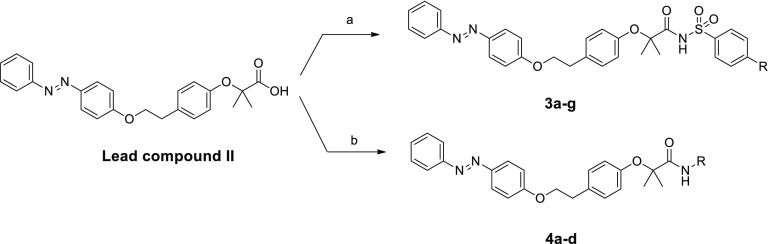
An agonist-antagonist switching strategy was performed to discover novel PPARα antagonists. Phenyldiazenyl derivatives of fibrates were developed, bearing sulfonimide or amide functional groups. A second series of compounds was synthesized, replacing the phenyldiazenyl moiety with amide or urea portions. Final compounds were screened by transactivation assay, showing good PPARα antagonism and selectivity at submicromolar concentrations. When tested in cancer cell models expressing PPARα, selected derivatives induced marked effects on cell viability. Notably, , , and displayed remarkable antiproliferative effects in two paraganglioma cell lines, with CC lower than commercial PPARα antagonist GW6471 and a negligible toxicity on normal fibroblast cells. Docking studies were also performed to elucidate the binding mode of these compounds and to help interpretation of SAR data.
采用激动剂-拮抗剂转换策略来发现新型PPARα拮抗剂。开发了带有磺酰亚胺或酰胺官能团的贝特类苯基重氮基衍生物。合成了第二系列化合物,用酰胺或脲部分取代苯基重氮基部分。通过反式激活试验对最终化合物进行筛选,结果表明这些化合物在亚微摩尔浓度下具有良好的PPARα拮抗作用和选择性。当在表达PPARα的癌细胞模型中进行测试时,所选衍生物对细胞活力产生显著影响。值得注意的是,[此处原文缺失具体化合物名称]在两种副神经节瘤细胞系中表现出显著的抗增殖作用,其半数抑制浓度低于市售PPARα拮抗剂GW6471,并且对正常成纤维细胞的毒性可忽略不计。还进行了对接研究以阐明这些化合物的结合模式,并有助于解释构效关系数据。