Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
Department of Veterans Affairs Medical Center, Pittsburgh, Pennsylvania.
JAMA Neurol. 2020 Aug 1;77(8):982-991. doi: 10.1001/jamaneurol.2020.1264.
An unmet need remains for safe and efficacious treatments for Duchenne muscular dystrophy (DMD). To date, there are limited agents available that address the underlying cause of the disease.
To evaluate the safety, tolerability, and efficacy of viltolarsen, a novel antisense oligonucleotide, in participants with DMD amenable to exon 53 skipping.
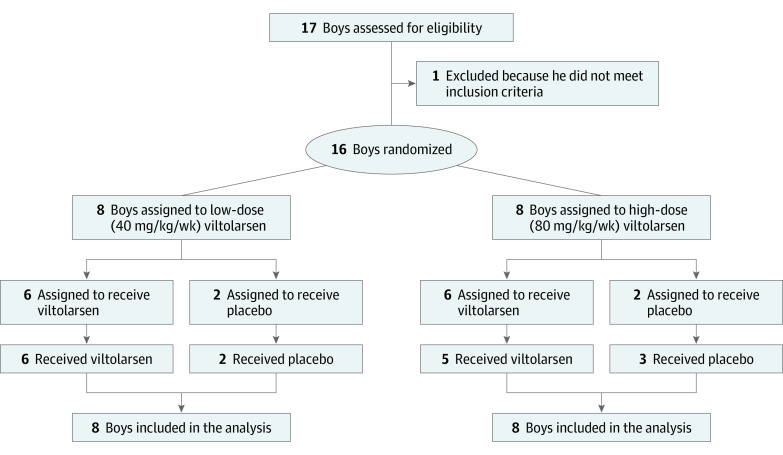
DESIGN, SETTING, AND PARTICIPANTS: This phase 2 study was a 4-week randomized clinical trial for safety followed by a 20-week open-label treatment period of patients aged 4 to 9 years with DMD amenable to exon 53 skipping. To enroll 16 participants, with 8 participants in each of the 2 dose cohorts, 17 participants were screened. Study enrollment occurred between December 16, 2016, and August 17, 2017, at sites in the US and Canada. Data were collected from December 2016 to February 2018, and data were analyzed from April 2018 to May 2019.
Participants received 40 mg/kg (low dose) or 80 mg/kg (high dose) of viltolarsen administered by weekly intravenous infusion.
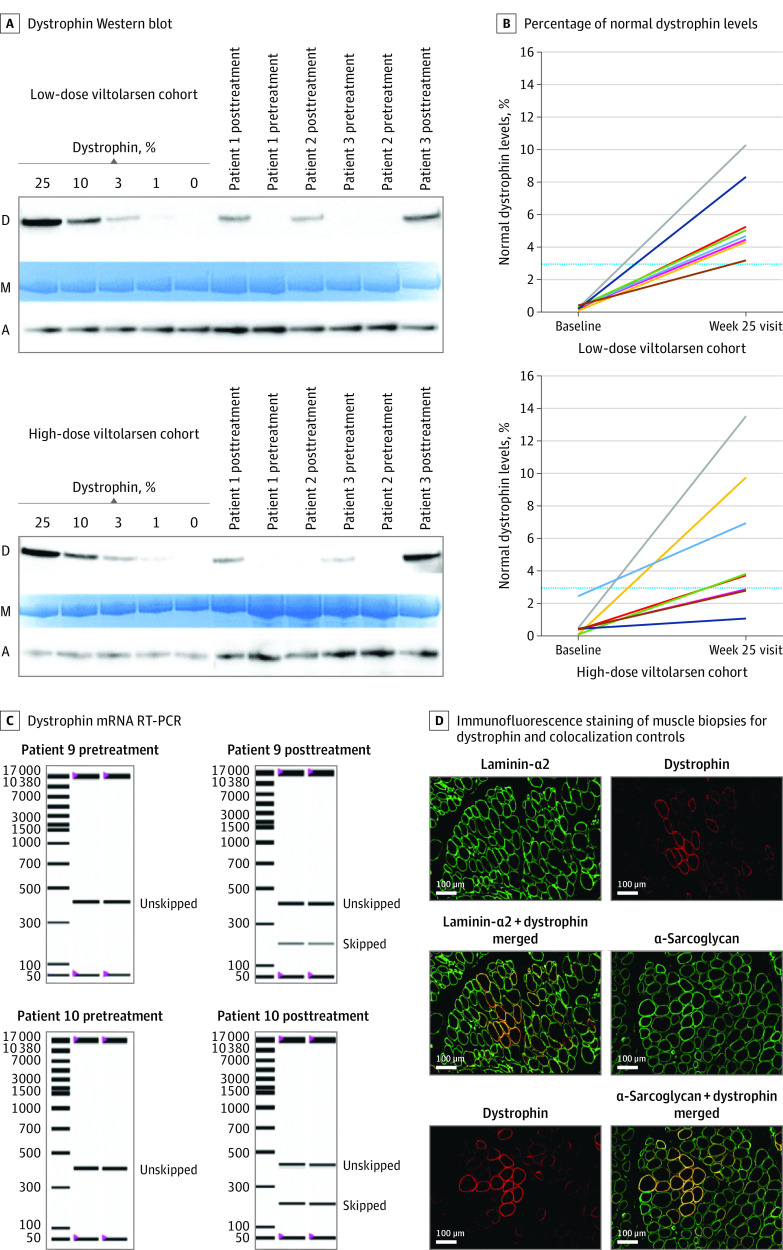
Primary outcomes of the trial included safety, tolerability, and de novo dystrophin protein production measured by Western blot in participants' biceps muscles. Secondary outcomes included additional assessments of dystrophin mRNA and protein production as well as clinical muscle strength and function.
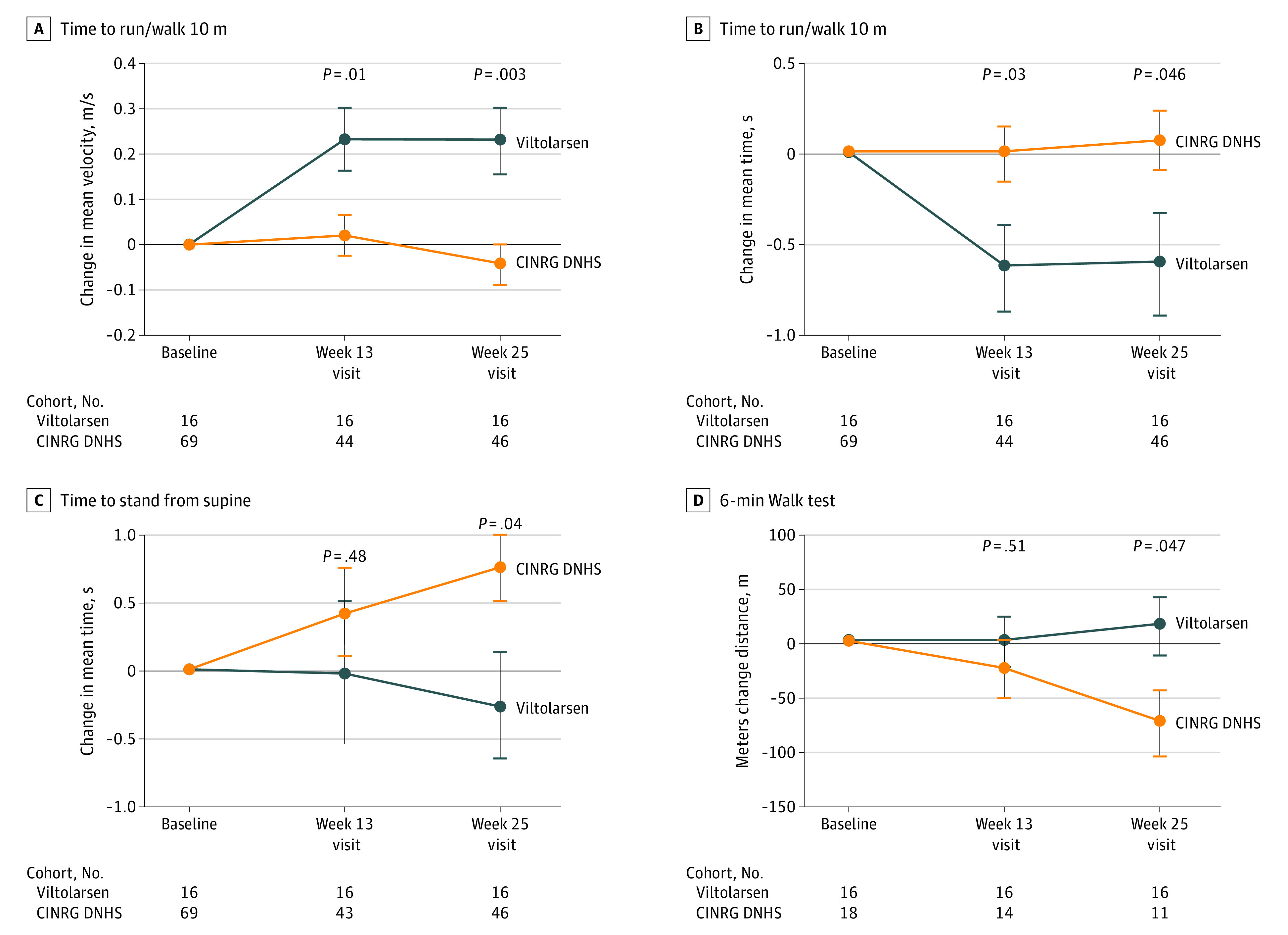
Of the 16 included boys with DMD, 15 (94%) were white, and the mean (SD) age was 7.4 (1.8) years. After 20 to 24 weeks of treatment, significant drug-induced dystrophin production was seen in both viltolarsen dose cohorts (40 mg/kg per week: mean [range] 5.7% [3.2-10.3] of normal; 80 mg/kg per week: mean [range] 5.9% [1.1-14.4] of normal). Viltolarsen was well tolerated; no treatment-emergent adverse events required dose reduction, interruption, or discontinuation of the study drug. No serious adverse events or deaths occurred during the study. Compared with 65 age-matched and treatment-matched natural history controls, all 16 participants treated with viltolarsen showed significant improvements in timed function tests from baseline, including time to stand from supine (viltolarsen: -0.19 s; control: 0.66 s), time to run/walk 10 m (viltolarsen: 0.23 m/s; control: -0.04 m/s), and 6-minute walk test (viltolarsen: 28.9 m; control: -65.3 m) at the week 25 visit.
Systemic treatment of participants with DMD with viltolarsen induced de novo dystrophin production, and clinical improvement of timed function tests was observed.
ClinicalTrials.gov Identifier: NCT02740972.
重要性:目前仍需要安全有效的治疗药物来治疗杜氏肌营养不良症(DMD)。到目前为止,能够针对疾病根本原因的药物非常有限。
目的:评估新型反义寡核苷酸药物 viltolarsen 对于适合外显子 53 跳跃的 DMD 患者的安全性、耐受性和疗效。
设计、环境和参与者:这是一项为期 4 周的安全性随机临床试验,随后进行了 20 周的开放性治疗期,纳入了年龄在 4 至 9 岁之间、适合外显子 53 跳跃的 DMD 患者。为了招募 16 名参与者(每组 8 名),共筛选了 17 名参与者。研究于 2016 年 12 月 16 日至 2017 年 8 月 17 日在美国和加拿大的多个地点进行。数据收集于 2016 年 12 月至 2018 年 2 月,数据分析于 2019 年 4 月至 5 月进行。
干预措施:参与者接受 40mg/kg(低剂量)或 80mg/kg(高剂量)的 viltolarsen 每周静脉输注。
主要结果和测量指标:试验的主要结局包括安全性、耐受性和参与者肱二头肌中通过 Western blot 测量的新产生的抗肌萎缩蛋白。次要结局包括对肌萎缩蛋白 mRNA 和蛋白产生的额外评估以及临床肌肉力量和功能的评估。
结果:在纳入的 16 名患有 DMD 的男孩中,有 15 名(94%)为白人,平均(标准差)年龄为 7.4(1.8)岁。经过 20 至 24 周的治疗后,两个 viltolarsen 剂量组均观察到药物诱导的抗肌萎缩蛋白显著产生(40mg/kg/周:平均[范围]为正常的 5.7%[3.2-10.3];80mg/kg/周:平均[范围]为正常的 5.9%[1.1-14.4])。viltolarsen 耐受性良好;没有因治疗引起的不良事件需要减少、中断或停止研究药物。研究期间没有发生严重不良事件或死亡。与 65 名年龄匹配和治疗匹配的自然病史对照者相比,所有接受 viltolarsen 治疗的 16 名参与者在从基线开始的计时功能测试中均表现出显著改善,包括从仰卧位到站立的时间(viltolarsen:-0.19s;对照组:0.66s)、从仰卧位到跑步/行走 10m 的时间(viltolarsen:0.23m/s;对照组:-0.04m/s)和 6 分钟步行测试(viltolarsen:28.9m;对照组:-65.3m)在第 25 周就诊时。
结论和相关性:全身性治疗 DMD 患者的 viltolarsen 诱导了新产生的抗肌萎缩蛋白,并且观察到计时功能测试的临床改善。
试验注册:ClinicalTrials.gov 标识符:NCT02740972。