Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Analytical and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA.
Nature. 2020 May;581(7809):452-458. doi: 10.1038/s41586-020-2329-2. Epub 2020 May 27.
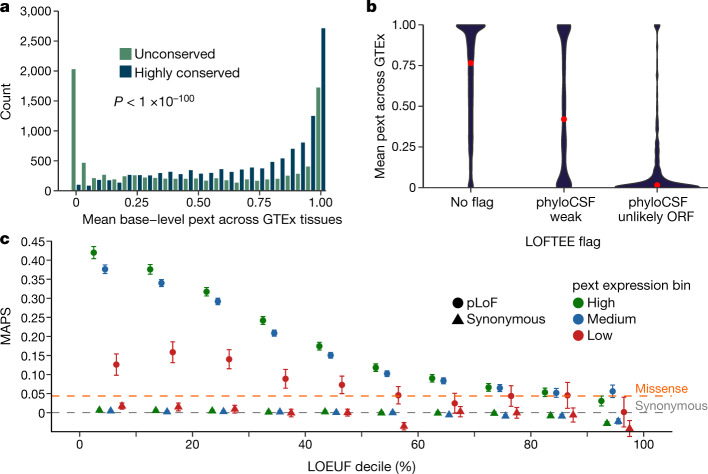
The acceleration of DNA sequencing in samples from patients and population studies has resulted in extensive catalogues of human genetic variation, but the interpretation of rare genetic variants remains problematic. A notable example of this challenge is the existence of disruptive variants in dosage-sensitive disease genes, even in apparently healthy individuals. Here, by manual curation of putative loss-of-function (pLoF) variants in haploinsufficient disease genes in the Genome Aggregation Database (gnomAD), we show that one explanation for this paradox involves alternative splicing of mRNA, which allows exons of a gene to be expressed at varying levels across different cell types. Currently, no existing annotation tool systematically incorporates information about exon expression into the interpretation of variants. We develop a transcript-level annotation metric known as the 'proportion expressed across transcripts', which quantifies isoform expression for variants. We calculate this metric using 11,706 tissue samples from the Genotype Tissue Expression (GTEx) project and show that it can differentiate between weakly and highly evolutionarily conserved exons, a proxy for functional importance. We demonstrate that expression-based annotation selectively filters 22.8% of falsely annotated pLoF variants found in haploinsufficient disease genes in gnomAD, while removing less than 4% of high-confidence pathogenic variants in the same genes. Finally, we apply our expression filter to the analysis of de novo variants in patients with autism spectrum disorder and intellectual disability or developmental disorders to show that pLoF variants in weakly expressed regions have similar effect sizes to those of synonymous variants, whereas pLoF variants in highly expressed exons are most strongly enriched among cases. Our annotation is fast, flexible and generalizable, making it possible for any variant file to be annotated with any isoform expression dataset, and will be valuable for the genetic diagnosis of rare diseases, the analysis of rare variant burden in complex disorders, and the curation and prioritization of variants in recall-by-genotype studies.
DNA 测序在患者和人群研究样本中的加速发展产生了广泛的人类遗传变异目录,但对罕见遗传变异的解释仍然存在问题。这一挑战的一个显著例子是,即使在明显健康的个体中,剂量敏感疾病基因中的破坏性变异也存在。在这里,通过对基因组聚集数据库(gnomAD)中部分功能丧失(pLoF)变体的人工筛选,我们发现,这种悖论的一个解释涉及 mRNA 的选择性剪接,它允许一个基因的外显子在不同细胞类型中以不同的水平表达。目前,没有现有的注释工具系统地将外显子表达信息纳入变异的解释中。我们开发了一种称为“跨转录体表达比例”的转录水平注释度量标准,用于量化变体的异构体表达。我们使用来自基因型组织表达(GTEx)项目的 11706 个组织样本计算了这个度量标准,并表明它可以区分弱和高度进化保守的外显子,这是功能重要性的一个代理。我们证明,基于表达的注释选择性地筛选了 gnomAD 中部分功能丧失疾病基因中假注释的 pLoF 变体的 22.8%,而在同一基因中去除的高可信度致病性变体不到 4%。最后,我们将我们的表达筛选应用于自闭症谱系障碍和智力残疾或发育障碍患者的新生变体分析,以表明在弱表达区域中的 pLoF 变体与同义变体具有相似的效应大小,而在高表达外显子中的 pLoF 变体在病例中富集程度最高。我们的注释快速、灵活且具有通用性,使得任何变体文件都可以用任何异构体表达数据集进行注释,这对于罕见疾病的遗传诊断、复杂疾病中罕见变体负担的分析以及召回基因型研究中变体的整理和优先级排序都将具有重要价值。