Funk Stephan Michael, Guedaoura Sonya, Juras Rytis, Raziq Absul, Landolsi Faouzi, Luís Cristina, Martínez Amparo Martínez, Musa Mayaki Abubakar, Mujica Fernando, Oom Maria do Mar, Ouragh Lahoussine, Stranger Yves-Marie, Vega-Pla Jose Luis, Cothran Ernest Gus
Centro de Excelencia de Modelación y Computación Científica Universidad de La Frontera Temuco Chile.
Nature Heritage St. Lawrence UK.
Ecol Evol. 2020 Apr 12;10(10):4261-4279. doi: 10.1002/ece3.6195. eCollection 2020 May.
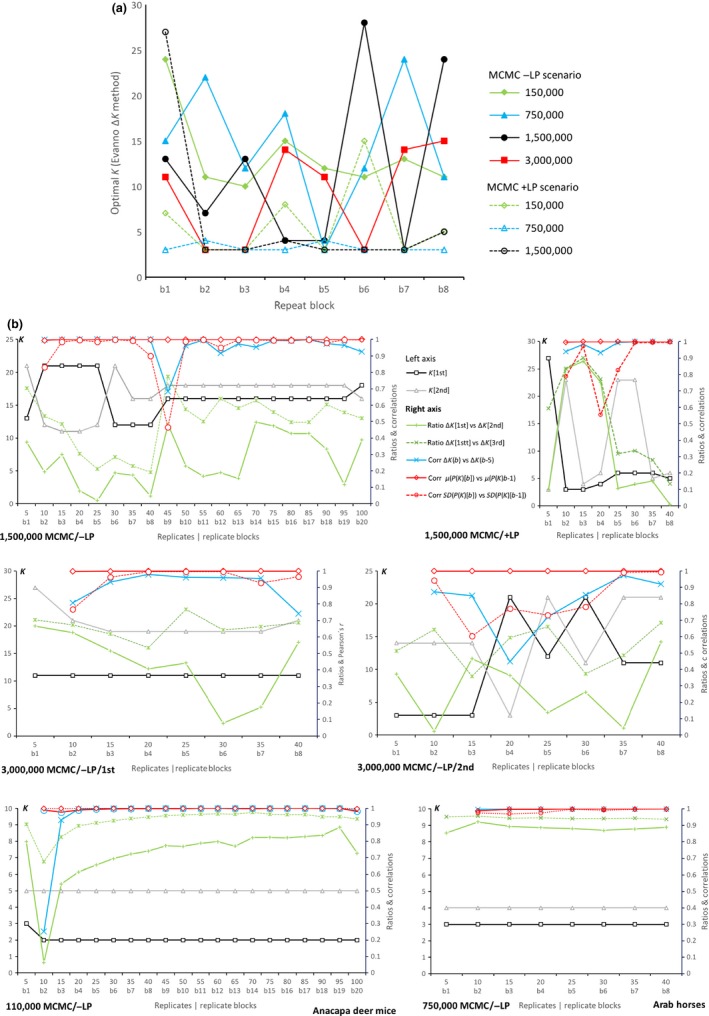
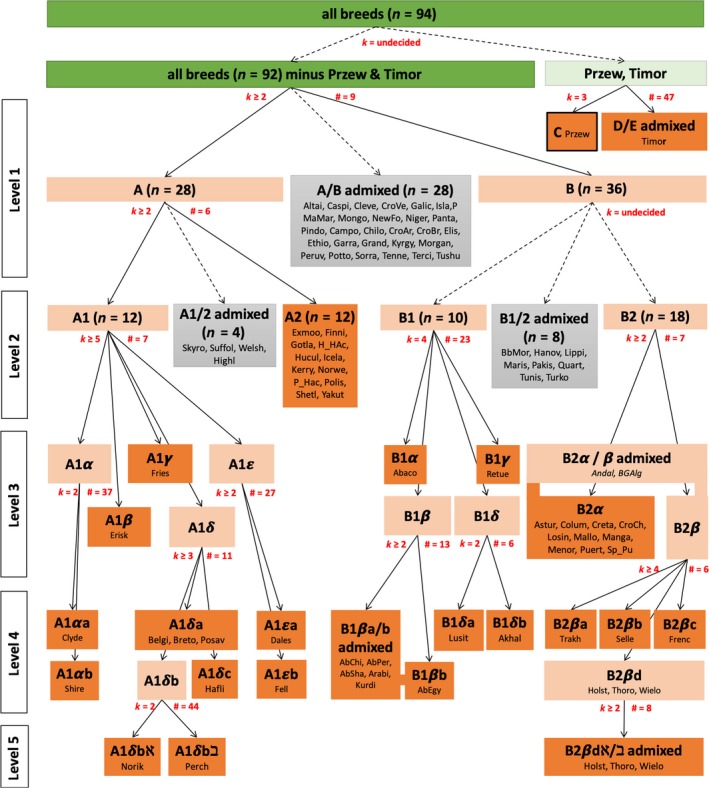
STRUCTURE remains the most applied software aimed at recovering the true, but unknown, population structure from microsatellite or other genetic markers. About 30% of structure-based studies could not be reproduced (, 21, 2012, 4925). Here we use a large set of data from 2,323 horses from 93 domestic breeds plus the Przewalski horse, typed at 15 microsatellites, to evaluate how program settings impact the estimation of the optimal number of population clusters that best describe the observed data. Domestic horses are suited as a test case as there is extensive background knowledge on the history of many breeds and extensive phylogenetic analyses. Different methods based on different genetic assumptions and statistical procedures (dapc, flock, PCoA, and structure with different run scenarios) all revealed general, broad-scale breed relationships that largely reflect known breed histories but diverged how they characterized small-scale patterns. structure failed to consistently identify using the most widespread approach, the Δ method, despite very large numbers of MCMC iterations (3,000,000) and replicates (100). The interpretation of breed structure over increasing numbers of , without assuming a , was consistent with known breed histories. The over-reliance on should be replaced by a qualitative description of clustering over increasing , which is scientifically more honest and has the advantage of being much faster and less computer intensive as lower numbers of MCMC iterations and repetitions suffice for stable results. Very large data sets are highly challenging for cluster analyses, especially when populations with complex genetic histories are investigated.
STRUCTURE仍然是最常用的软件,旨在从微卫星或其他遗传标记中恢复真实但未知的群体结构。约30%基于STRUCTURE的研究无法被重复(,2012年,4925)。在此,我们使用来自93个家养品种的2323匹马以及普氏野马的大量数据,这些数据在15个微卫星上进行了分型,以评估程序设置如何影响对最能描述观测数据的群体簇最佳数量的估计。家养马适合作为测试案例,因为许多品种的历史有广泛的背景知识以及广泛的系统发育分析。基于不同遗传假设和统计程序的不同方法(dapc、flock、主坐标分析以及不同运行场景的STRUCTURE)都揭示了大致反映已知品种历史的总体、大规模品种关系,但在如何刻画小规模模式方面存在分歧。尽管进行了非常大量的马尔可夫链蒙特卡罗迭代(3000000)和重复(100次),STRUCTURE仍未能使用最广泛的方法——Δ方法一致地识别。在不假设的情况下,随着的增加对品种结构的解释与已知品种历史一致。对的过度依赖应该被对随着增加的聚类的定性描述所取代,这在科学上更诚实,并且具有更快且计算强度更低的优点,因为较少数量的马尔可夫链蒙特卡罗迭代和重复就足以获得稳定结果。非常大的数据集对聚类分析极具挑战性,尤其是在研究具有复杂遗传历史的群体时。