Jungreis Irwin, Sealfon Rachel, Kellis Manolis
MIT Computer Science and Artificial Intelligence Laboratory, Cambridge, MA.
Broad Institute of MIT and Harvard, Cambridge, MA.
bioRxiv. 2020 Sep 2:2020.06.02.130955. doi: 10.1101/2020.06.02.130955.
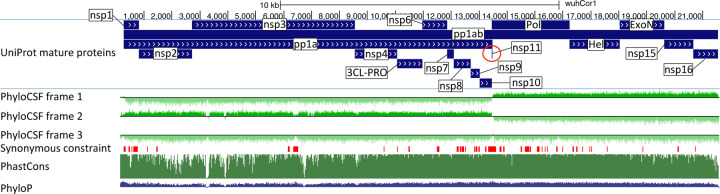
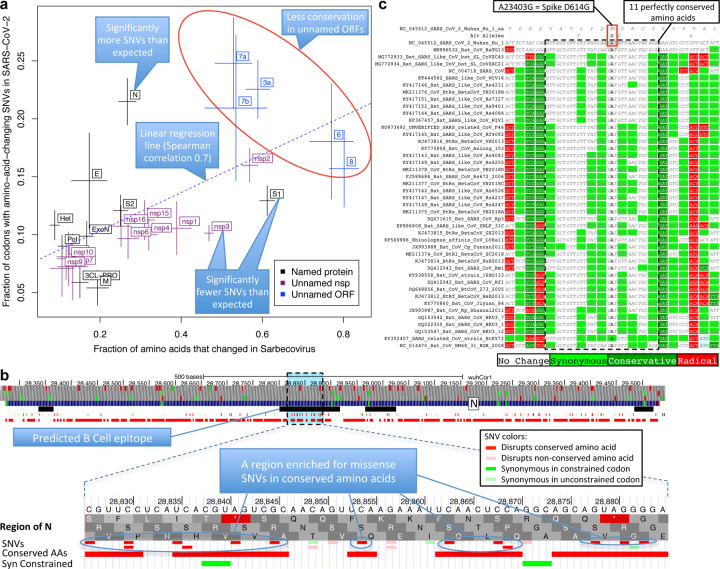
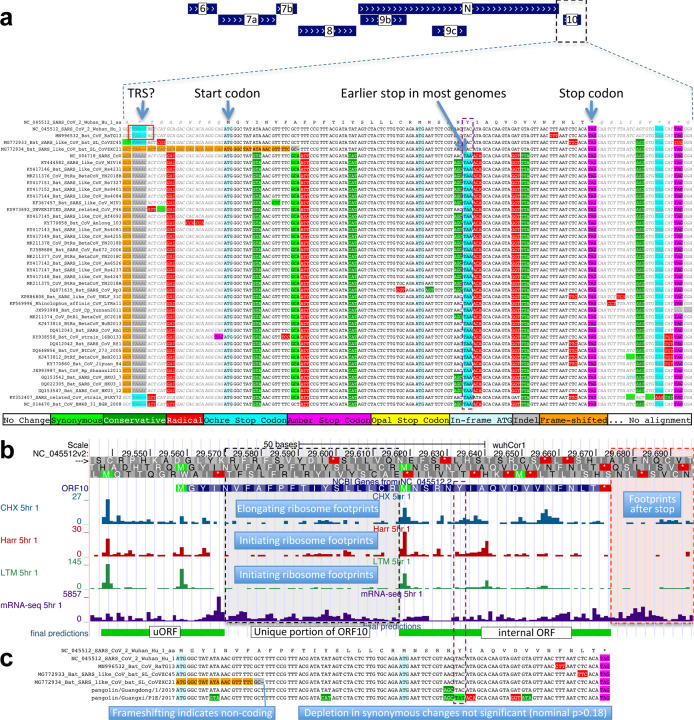
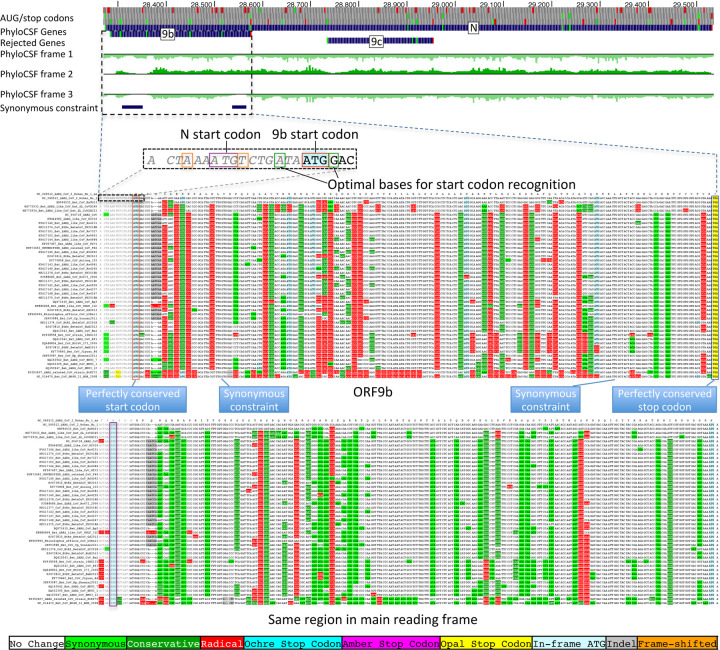
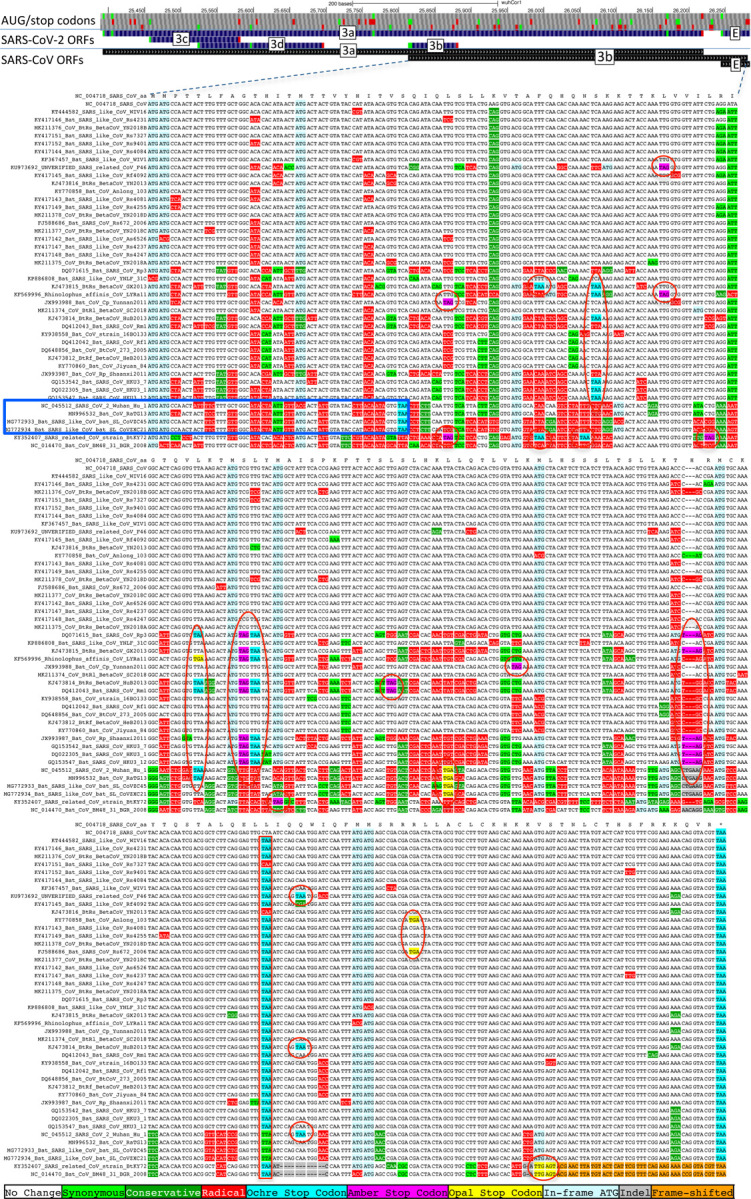
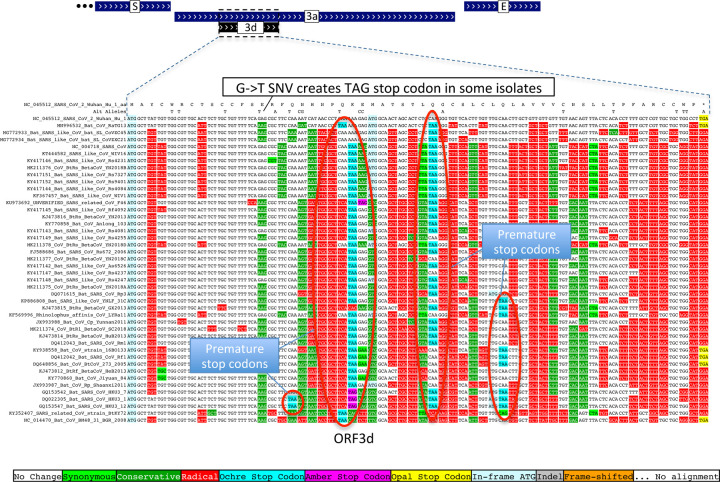
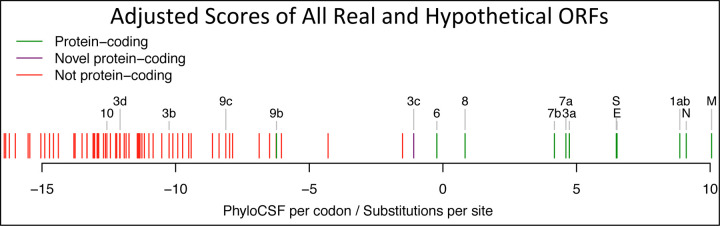
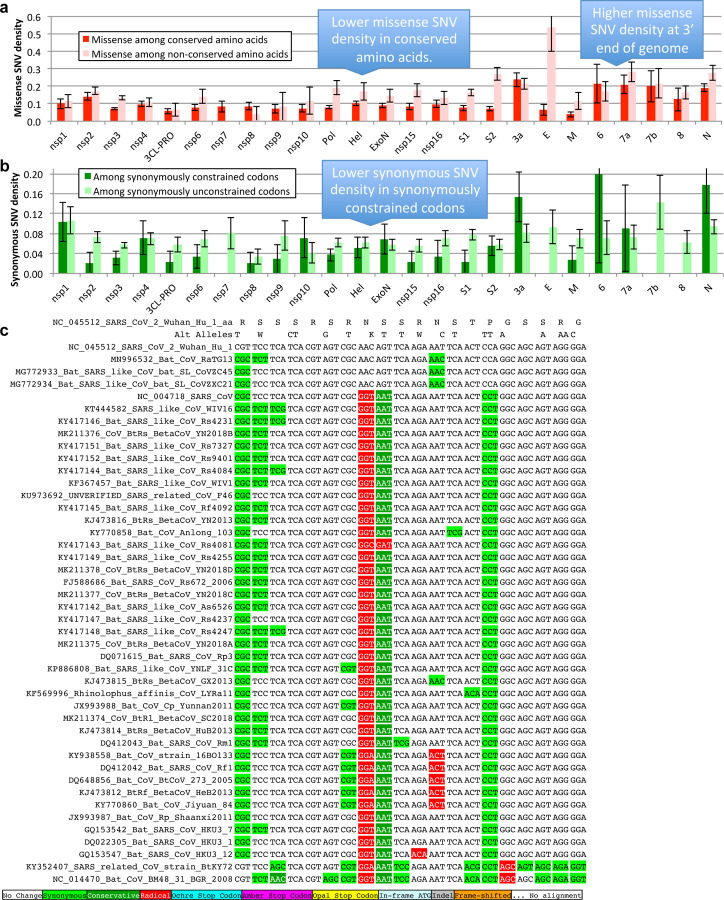
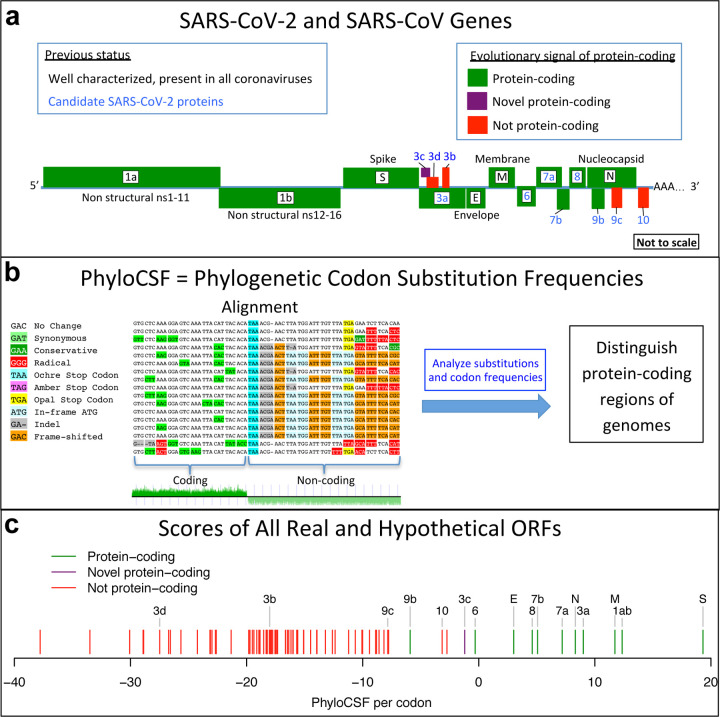
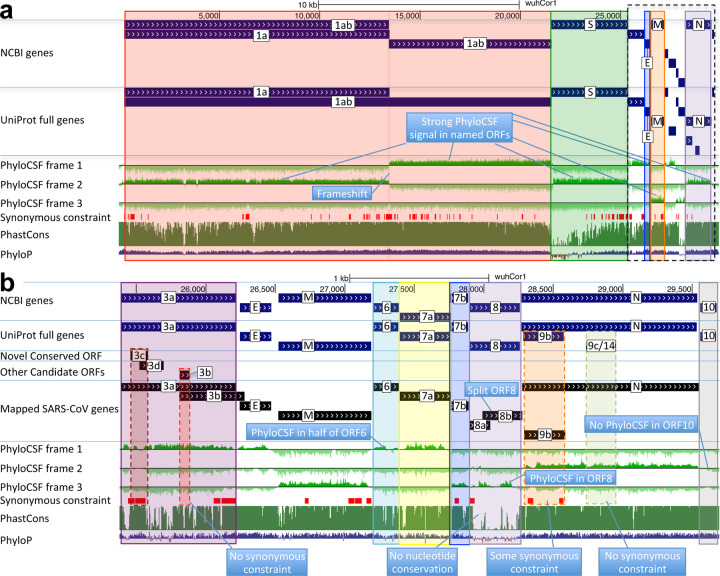
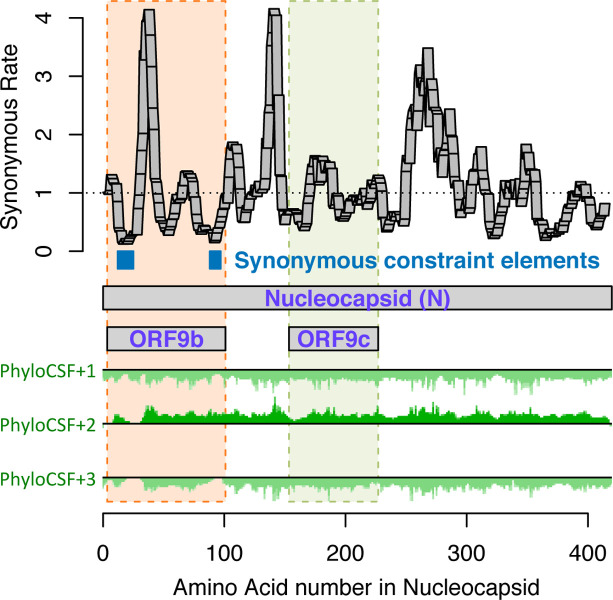
Despite its overwhelming clinical importance, the SARS-CoV-2 gene set remains unresolved, hindering dissection of COVID-19 biology. Here, we use comparative genomics to provide a high-confidence protein-coding gene set, characterize protein-level and nucleotide-level evolutionary constraint, and prioritize functional mutations from the ongoing COVID-19 pandemic. We select 44 complete Sarbecovirus genomes at evolutionary distances ideally-suited for protein-coding and non-coding element identification, create whole-genome alignments, and quantify protein-coding evolutionary signatures and overlapping constraint. We find strong protein-coding signatures for all named genes and for 3a, 6, 7a, 7b, 8, 9b, and also ORF3c, a novel alternate-frame gene. By contrast, ORF10, and overlapping-ORFs 9c, 3b, and 3d lack protein-coding signatures or convincing experimental evidence and are not protein-coding. Furthermore, we show no other protein-coding genes remain to be discovered. Cross-strain and within-strain evolutionary pressures largely agree at the gene, amino-acid, and nucleotide levels, with some notable exceptions, including fewer-than-expected mutations in nsp3 and Spike subunit S1, and more-than-expected mutations in Nucleocapsid. The latter also shows a cluster of amino-acid-changing variants in otherwise-conserved residues in a predicted B-cell epitope, which may indicate positive selection for immune avoidance. Several Spike-protein mutations, including D614G, which has been associated with increased transmission, disrupt otherwise-perfectly-conserved amino acids, and could be novel adaptations to human hosts. The resulting high-confidence gene set and evolutionary-history annotations provide valuable resources and insights on COVID-19 biology, mutations, and evolution.
尽管严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的基因组具有极其重要的临床意义,但其基因集仍未明确,这阻碍了对2019冠状病毒病(COVID-19)生物学特性的剖析。在此,我们利用比较基因组学来提供一个高可信度的蛋白质编码基因集,表征蛋白质水平和核苷酸水平的进化限制,并对正在肆虐的COVID-19大流行中的功能性突变进行优先级排序。我们选择了44个进化距离理想的完整Sarbecovirus属基因组,以用于蛋白质编码和非编码元件的识别,创建全基因组比对,并量化蛋白质编码的进化特征和重叠限制。我们发现所有已命名基因以及3a、6、7a、7b、8、9b还有一个新的可变阅读框基因ORF3c都有很强的蛋白质编码特征。相比之下,ORF10以及重叠阅读框9c、3b和3d缺乏蛋白质编码特征或令人信服的实验证据,并非蛋白质编码基因。此外,我们表明没有其他蛋白质编码基因有待发现。跨毒株和毒株内的进化压力在基因、氨基酸和核苷酸水平上基本一致,但也有一些显著例外,包括非结构蛋白3(nsp3)和刺突蛋白亚基S1中的突变少于预期,而核衣壳蛋白中的突变多于预期。后者在预测的B细胞表位中其他保守残基处还显示出一组氨基酸变化的变体,这可能表明存在免疫逃避的正选择。包括与传播增加有关的D614G在内的几个刺突蛋白突变破坏了原本完全保守的氨基酸,可能是对人类宿主的新适应。由此产生的高可信度基因集和进化历史注释为COVID-19的生物学特性(包括突变和进化)提供了宝贵的资源和见解。